VISUALIZING BIOLOGICAL DATA VIZBI
Register for VIZBI 2026
Posters
Thumbnails:
List:
Year:
Category:
Session:
Poster:
Getting poster data...

Clustering biomolecular structures by residue contact similarity
João PGLM Rodrigues, Mikaël Trellet, Christophe Schmitz, Adrien SJ Melquiond, Alexandre MJJ Bonvin (Padualaan 8, Utrecht, The Netherlands)
Comparing and clustering three-dimensional protein structures is a commonly employed end-game strategy in protein structure prediction. While the root mean square deviation of atomic coordinates (rmsd) between pairs of structures underlies most of these clustering methods, it becomes slow to compute for larger datasets, and also inaccurate for larger systems. We present a similarity measure based on atomic contacts that allows clustering in a much shorter time (~100-fold reduction) than rmsd methods without compromising accuracy. Our measure, the fraction of common contacts, is able to deal effortlessly with symmetrical assemblies that would otherwise require lengthy iterative rmsd calculations. It also handles different types of molecules such as proteins, nucleic acids and small ligands. Overall, our method offers an all-round computationally cheap solution to the problem of structural clustering in protein docking.
Dr Jon Heras (Equinox Graphics Ltd, 85 Bishops Road, Cambridge, CB2 9NQ, United Kingdom)
0 2026 Proteins A

Daniel Rice, Swaathi Kandasaamy, Minjoon Kim, Maria Martin ( EMBL- European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridgeshire, CB10 1SD, UK)
1 2026 Proteins A

Elira Sabouri, Kay Nieselt (InterfakultÀres Institut fÌr Biomedizinische Informatik (IBMI))
2 2026 Genomes A

Irina Titkova (Show-cell)
3 2026 Proteins A

D. Sehnal, T. Slanináková, Z. Charlop-Powers, V. Doshchenko, A. S. Rose, A. Midlik, A. SekuÅa, N. Fonseca, K. L. Morris, S. K. Burley, S. Velankar, J. Fleming, B. Vallat, L. Autin (Masaryk University, Brno, Czechia; PDBe, Hinxton, UK; RCSB, La Jolla, USA; EMDB, Hinxton, UK)
4 2026 Proteins B

Josh Hawley (Golgi Graphics, 71-75 Shelton Street, Covent Garden, London, England, WC2H 9JQ)
5 2026 Cells B

Michael K. Jones, Charlotte Capitanchik, Sam Ireland, Alex Harston, Martin Husbyn, Urska Janjos, Benjamin Blencowe, Jernej Ule (5 Cutcombe Rd, London UK )
6 2026 Transcripts B

Sophie den Hartog, Emelie Brodrick, Gaspar Jekely, Vengamanaidu Modepalli & Elizabeth Williams (University of Exeter, Exeter, England; the Marine Biological Association, Plymouth, England)
7 2026 Cells C

Katherine A. Wolcott, Edward L. Stanley, Arthur Porto (Florida Museum of Natural History, Gainesville, FL (USA))
8 2026 Organisms C


Omar Mena¹, UroÅ¡ Å majdek², Weiping Zhang³, Piao Yu¹, Tobias KleinâŽ, Stefan Arold¹, Sai Li³, Ivan Viola¹, Ciril Bohak¹➎² (¹ KAUST, ² University of Ljubljana, ³ Tsinghua University, ⎠Nanographics)
10 2026 Other D

Michael Krone, Anna Sterzik, Kai Lawonn (Stuttgart Technical University of Applied Sciences, Stuttgart, Germany; University of Jena, Jena, Germany)
11 2026 Proteins D

Vera Williams (Institut Pasteur, Paris, France)
12 2026 Proteins D

Jim Procter, Renia Correya, Ben Soares, Mateusz Warowny, Mungo Carstairs, Thomas van Aalten, Khadija Jabeen, Suzanne Duce, Geoff Barton (Faculty of Life Sciences, University of Dundee, Scotland, UK.)
13 2026 Proteins D

Z. Lyu, E. Wang, M. Yang, F. Mo, F. Wang, W. Zhao, Z. Guan, H. Cao, Y. Jiang, C. Cui, Z. Sun, Z. Liu, A. Abdullah, C. Zhang, A. C. Knoll, P. Lio (Xidian Univ.; Anhui Univ. of Science and Technology; Univ. of Electronic Science and Technology of China; Tianjin Univ.; Technical Univ. of Munich; Univ. of Cambridge; Univ. of Oxford)
14 2026 Genomes A

Kristen Browne, Meghan McCarthy, and Darrell E. Hurt (5601 Fishers Lane, Rockville, Maryland)
15 2026 Proteins A

Therese Hoang1,2, Miriam Yeung1,2, Difei Huang1, Chirag Manoj1, Katrina Karapanos1, Lauren Tjoeka1,2, Laura Kirby1, Sarah-Jane Dawson1,2, Stephen Wong1,2, Elizabeth Christie1,2, George Au-Yeung1,2 (1. Peter MacCallum Cancer Centre, Melbourne, Victoria, Australia 2. Sir Peter MacCallum Department of Oncology, The University of Melbourne, Parkville, Victoria, Australia )
16 2026 Genomes B

Hasan Balci, Augustin Luna (Computational Biology Branch, National Library of Medicine, NIH, Bethesda, MD, 20892, USA)
17 2026 Cells C
")
Seán OâDonoghue, James Procter, Bara Kozliková, Theodoros Soldatos, Christian Stolte (Magdalene College, University of Cambridge)
18 2026 Art & Biology Y

Josh Hawley (Golgi Graphics, 71-75 Shelton Street, Covent Garden, London, England, WC2H 9JQ)
19 2026 Art & Biology Y

Joost Bakker, Geurt van Hierden (Scicomvisuals BV, Amsterdam, The Netherlands)
20 2026 Art & Biology Y

Hafsa Malik (Federal Medical College, Hanna Road, G8/4, Islamabad, Pakistan )
21 2026 Art & Biology Y

Dr Jon Heras (Equinox Graphics Ltd, 85 Bishops Road, Cambridge, CB2 9NQ, United Kingdom)
22 2026 Art & Biology Y



Samuel Pantze (Center for Advanced Systems Understanding, Görlitz, Germany)
25 2026 Art & Biology Y

Leonora MartÃnez-Nuñez (Radiant Molecules Studio. Brisbane, QLD. Australia.)
26 2026 Art & Biology Y

Andrea Winterbottom, Jamie Allen, Stefano Giorgetti, Leanne Haggerty, Garth R Ilsley, Jon Keatley, John Tate, Bethany Flint, Sarah E Hunt, Robert D Finn, Andrew D Yates (European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge, CB10 1SD, United Kingdom)
27 2025 Genomes A

Daniela Volpatto, Simone Pernice, Sandro Gepiro Contaldo, Roberta Sirovich, Francesca Cordero, Marco Beccuti (University of Turin, Turin, Italy)
28 2025 Populations A

Derek W Wright, Laura Mojsiejczuk, Afrida Mukaddas, Joseph Hughes, Robert J Gifford, David L Roberston, Daniel Goldhill, Edward C Hutchinson (MRC-University of Glasgow Centre for Virus Research, Sir Michael Stoker Building, Garscube Campus, 464 Bearsden Road, Glasgow G61 1QH Scotland (UK))
29 2025 Genomes A

M Maria, S GuionniÚre, N Dacquay, C Plateau-Holleville, V Guillaume, V Larroque, J Lardé, Y Naimi, JP Piquemal, G Levieux, N Lagarde, S Mérillou, M Montes (CNAM, Paris, France; Université de Limoges, Limoges, France; Sorbonne Université, PAris, France; Qubit Pharmaceuticals, Paris, France; IUF, Paris, France)
30 2025 Proteins A

Dr Jon Heras (Equinox Graphics Ltd, 85 Bishops Road, Cambridge, CB2 9NQ, United Kingdom)
31 2025 Proteins A

Adam Midlik, Daniel Rice, Marcelo Afonso, Sreenath Nair, Jennifer Flemming, Sameer Velankar (European Molecular Biology Laboratory, European Bioinformatics Institute, Hinxton, Cambridge CB10 1SD, UK)
32 2025 Proteins A

Megan Gozzard (Wellcome Sanger Institute)
33 2025 Other A

Carolin Willner, Jennifer Sapia, Bianca M. Esch, Pia ErdbrÌgger, Sergej Limar, Sebastian Eising, Carolin Körner, Stefano Vanni, Florian Fröhlich (OsnabrÌck University; Department of Biology/Chemistry; Bioanalytical Chemistry section; 49076 OsnabrÌck, Germany; )
34 2025 Proteins A

Stefan Manolache, Dr. Hajk-Georg Drost (University of Dundee, Dundee, UK)
35 2025 Transcripts A

Irina Titkova, Tanya Soliman (Barts Cancer Institute, QMUL, John Vane Science Centre, Charterhouse Square, London EC1M 6BQ, UK)
36 2025 Proteins B

Véronique Hourdel, Pierre Lechat, Aurelia Kwasiborski, Rémi Vincent, Lucie Cappuccio, Valérie Caro and Jean-Claude Manuguerra (Institut Pasteur 28 rue du docteur Roux, 75015 PARIS, FRANCE)
37 2025 Genomes B

Sylwia Czach, Katarzyna Walczewska-Szewc (Nicolaus Copernicus University, Torun, Poland)
38 2025 Proteins B

Samuel Pantze, Matthew McGinity, Ulrik GÌnther (Center for Advanced Systems Understanding, Görlitz, Germany; Helmholtz-Zentrum Dresden-Rossendorf, Germany; IXLAB Technische UniversitÀt Dresden, Germany)
39 2025 Cells B

Neli Fonseca, Zhe Wang, Jack Turner, Lucas Carrijo, Miao Ma, Minoosadat Tayebinia, Kyle Morris (European Bioinformatics Institute, Hinxton, UK)
40 2025 Genomes B

Jessica Heebner, Holly Peterson (Thermo Fisher Scientific, Hillsboro, OR, USA)
41 2025 Cells B

Oktawia Korcz, Tomasz WÅodarski (Department of Bioinformatics, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, ul. PawiÅskiego 5A, 02-106 Warsaw, Poland)
42 2025 Proteins B

Maria Kiourlappou1 , Peter Todd1, Yaxuan Kong2, Jayesh Hire1 , Martin Sergeant3, Stefan Zohren2, Jim Hughes3, Stephen Taylor1 (1Centre of Human Genetics, University of Oxford, 2Department of Engineering, University of Oxford, 3Weatherall Institute of Molecular Medicine, University of Oxford)
43 2025 Transcripts B

E. Gaibor, C. Rorden, R. Pienaar, D. Haehn (University of Massachusetts Boston)
44 2025 Organisms B

Andrew McCluskey, Thomas D. Otto (University of Glasgow, Glasgow, UK; Université de Montpellier, Montpellier, France)
45 2025 Transcripts B

Swaathi Kandasaamy, Daniel Rice, Aurélien Luciani, Gustavo Salazar, Matthias Blum, Adam Midlik, Marcelo Querino, Maria Martin (EMBL - European Bioinformatics Institute)
46 2025 Proteins C

David Sehnal, Adam Midlik, Sebastian Bittrich, Terézia Slanináková, Tomáš RaÄek, Jennifer R. Fleming, Sreenath Nair, Sameer Velankar, Stephen K. Burley, Jasmine Y. Young, Brinda Vallat (PDBe, Hinxton, UK; RCSB, La Jolla, California, USA; NCBR, Masaryk University, Brno, Czech Republic)
47 2025 Proteins C

Konstantinos Alexandrou, Buse Isbilir, Andriko von KÃŒgelgen, Tanmay A.M. Bharat, Shraddha Nayak (MRC Laboratory of Molecular Biology, Cambridge Biomedical Campus, Francis Crick Ave, Trumpington, Cambridge CB2 0QH)
48 2025 Genomes C

Laura Olivares Boldú (Wellcome Connecting Science, Wellcome Genome Campus, Hinxton, UK)
49 2025 Other C


Chisato Tsuji (Random42/University of Bristol/MRC Laboratory of Molecular Biology)
51 2025 Proteins C

Julia Patricia Schessner, Mikhail Lebedev, Magnus Schwörer, Patricia Skowronek, Matthias Mann (Max-Planck-Institute for Biochemistry, Martinsried, Germany)
52 2025 Proteins C

Muhammed Khawatmi, Shivprasad Jamdade, and Heba Sailem (King's College London)
53 2025 Genomes C

Blake A Sweeney, Holly McCann, et al, Anton I Petrov (EMBL-EBI, Wellcome Genome Campus, Hinxton, Cambridgeshire, CB10 1SD, UK)
54 2025 Other C

Grace Hsu and Andrew Catalano (Smart Biology Inc., Ottawa, ON, Canada)
55 2025 Proteins A

Gabor Beke, Milan Hucko, Lubos Klucar, Dana Cholujova, Jana Jakubikova (1Institute of Molecular Biology, Slovak Academy of Sciences, Bratislava, Slovakia; Cancer Research Institute, Biomedical Research Center, Slovak Academy of Sciences, Bratislava, Slovakia)
56 2025 Genomes A

Andrew C. Tapia, Jerzy W. Jaromczyk, Neil Moore, and Christopher L. Schardl (329 Rose St., Lexington, KY 40508)
57 2025 Populations B

VinÃcius A. Paiva¹, Douglas E. V. Pires², Gustavo C. Bressan¹, Sandro C. Izidoro³, Sabrina A. Silveira¹ (1. Universidade Federal de Viçosa, Brazil | 2. University of Melbourne, Australia | 3. Universidade Federal de Itajubá, Brazil)
58 2025 Proteins B

Lea C. von Soosten (HARBOR, Luruper Chaussee 149, 22761 Hamburg, Germany)
59 2025 Proteins C

Duygu Koldere Vilain (Designosome Scientific Visuals)
60 2025 Art & Biology Y

Joost M. Bakker (Scicomvisuals, Gillis van Ledenberchstraat 76, 1052VK Amsterdam, The Netherlands)
61 2025 Art & Biology Y

Samuel Pantze (Center for Advanced Systems Understanding, Görlitz, Germany; Helmholtz-Zentrum Dresden-Rossendorf, Germany)
62 2025 Art & Biology Y

Mark Danson, Laura Olivares Boldú (Wellcome Connecting Science, Wellcome Genome Campus, Hinxton, UK)
63 2025 Art & Biology Y

Dr Jon Heras (Equinox Graphics Ltd, 85 Bishops Road, Cambridge, CB2 9NQ, United Kingdom)
64 2025 Art & Biology Y

Jessica Heebner, Holly Peterson (Thermo Fisher Scientific, Hillsboro, OR, USA)
65 2025 Art & Biology Y

Beata Edyta Mierzwa (University of California San Diego, La Jolla, USA; Beata Science Art, San Diego, USA)
66 2025 Art & Biology Y

67 2025 Art & Biology Y

Heba Sailem (King's College London)
68 2025 Art & Biology Y


Daniel Villanueva Avalos (Brigham Young University, Provo, UT 84602)
70 2025 Art & Biology Y



Elfy Chiang (MRC Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge CB2 0QH, UK.)
73 2025 Art & Biology Y

Rachel Torrez, Jarrod S. Johnson, and Janet Iwasa (University of Utah, Department of Biochemistry)
74 2024 Genomes A

Ann S. Blevins, Danielle Callan, John Brestelli, Jay Humphrey, Dave Falke, Jeremy Myers, Elizabeth Harper, Ryan Doherty, Dan Beiting (3800 Spruce Street - Philadelphia, PA 19104)
75 2024 Genomes A

Alexandre Kouyoumdjian, Ondrej Strnad, Ciril Bohak, Dawar Khan, Anwar Haredy, Donggang Jia, Ivan Viola (KAUST, Saudi Arabia)
76 2024 Proteins A


Damiano Clementel, Alexander Monzon, Silvio Tosatto (University of Padua, Department fo Biomedical Sciences, Via Ugo Bassi, 58/B, 35142 Padova, Italy)
78 2024 Proteins A

Jesse Weller, Remo Rohs (University of Southern California, 1050 Childs Way, Los Angeles, CA 90089)
79 2024 Proteins A

Raktim Mitra, Ari S. Cohen, Remo Rohs (Department of Quantitative & Computational Biology, University of Southern California Los Angeles, CA 90089, USA)
80 2024 Other A

Elul T, Lew V, Khetani S (Touro University California, Vallejo CA)
81 2024 Cells A

Kristen M. Browne, Meghan C. McCarthy, Phillip Cruz, David Liou, Darrell E. Hurt (5601 Fishers Ln, Rockville, MD 20852)
82 2024 Proteins B

Yangyang Bai, Matt Jones, Janielle Cuala, Lynne Cherchia, Lauro Sebastian Ojeda, Scott E. Fraser, Thai V. Truong (1219 W. 27th street)
83 2024 Populations B

Jim Stanis, Wei Sun, Yuchun Tang, Yuchuan Qiao, Xinting Ge, Mara Mather, John M Ringman, Yonggang Shi (2025 Zonal Ave. Los Angeles, CA 90033)
84 2024 Organisms B

Katrin Rein (German Cancer Research Center, Heidelberg, Germany)
85 2024 Organisms B

Raktim Mitra, Jinsen Li, Jared Sagendorf, Yibei Jiang, Tsu-Pei Chiu, Cameron J. Glasscock, and Remo Rohs (University of Southern California Los Angeles, CA 90089, USA)
86 2024 Proteins B

John âScooterâ Morris, Tom Ferrin, Tom Goddard, Eric Pettersen, Greg Couch, Elaine Meng, Zach Pearson (UCSF, San Francisco, USA)
87 2024 Proteins B

Beata Edyta Mierzwa, Matthew Cooney (UC San Diego, La Jolla, USA)
88 2024 Other B

Jordy Homing Lam, Aiichiro Nakano and Vsevolod Katritch (University of Southern California)
89 2024 Proteins C

Maxime Maria, Simon Guionniere, Nicolas Dacquay, Valentin Guillaume, Cyprien Plateau-Holleville, Stéphane Mérillou, Yassine Naimi, Jean-Philip Piquemal, Guillaume Levieux, Matthieu Montes (XLIM, UMR CNRS7252, Université de Limoges, France; GBCM EA7528, CNAM, France; Qubit-Pharmaceuticals SAS; LCT, UMR CNRS7616, Sorbonne Université, France;CEDRIC EA4626, CNAM, France; IUF, France)
90 2024 Cells C

Tyler Ard, Michael S. Bienkowski, James Stanis, Caroline OâDriscoll, Arthur W. Toga (University of Southern California, Los Angeles, USA)
91 2024 Other C

Yibei Jiang, Raktim Mitra, Remo Rohs (1050 Childs Way, Los Angeles, CA 90089)
92 2024 Proteins C

Katy Borner et al. (Indiana University)
93 2024 Other C

Marina Murillo-Recio (Institute for Research in Biomedicine)
94 2024 Genomes C

Wen Lin, Nicholas Pudjarminta, Bryan Zhang, Helen Berman, Alex McDowell, Kate White (USC Bridge Institute, Los Angeles, USA; World Building Media Lab - School of Cinematics, University of Southern California)
95 2024 Proteins C

Anh Hang Mai Vo, Tien-Anh Vu ([1] Freelance Scietific Illustrator, Vinmec High Tech Center; [2] VNU University of Science)
96 2024 Proteins C

Eric Lu, Michelle Wei, Enrique Noriega, Sateesh Peri, Mihai Surdeanu, Guang Yao (University of Arizona, Tucson, USA)
97 2024 Genomes A

Susie Huget, Juan Jose Medina Reyes, Henning Hermjakob (EMBL-EBI, Wellcome Genome Campus, Hinxton, Cambridgeshire, CB10 1SD, UK)
98 2024 Genomes A

Aaron Watters (Flatiron Institute, New York, NY, USA)
99 2024 Organisms A

Philip Hubbard (Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA, USA)
100 2024 Other B
: An AI & NLP enabled approach.")
Piyush Kumar Singh, Theodoros Soldatos (SRH Heidelberg, Germany.)
101 2024 Other B

Mahsa Mofidi, Chris Edelmaier, Abhilash Sahoo, Sonya M. Hanson, Ehssan Nazockdast (Department of Applied Physical Sciences, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599)
102 2024 Proteins B

Eliot Ragueneau, Chuqiao Gong, Henning Hermjakob (EMBL-EBI, Cambridge, UK)
103 2024 Cells C

Vincenzo Costanzo1, Ludovica Altieri1,2, Patrizia Lavia1 ( (1) Institute of Molecular Biology and Pathology (IBPM), CNR National Research Council, Rome, Italy; (2) Department of Biology and Biotechnology âCharles Darwinâ, Sapienza University of Rome, Italy)
104 2024 Cells C

Marina A. Pak (Skolkovo Institute of Science and Technology, the territory of the Innovation Center âSkolkovo", Bolshoy Boulevard, 30, p.1, Moscow 121205, Russia)
105 2024 Proteins C

Seán O'Donoghue, James Procter, Barbora KozlÃková, Helen Berman (Bridge Institute, USC, USA)
106 2024 Art & Biology Y

Nicolas Romeo, Jonathan Jackson, Jasmin Imran Alsous (University of Chicago, Chicago, USA; Harvard University, Cambridge, USA; Flatiron Institute, New York, USA)
107 2024 Art & Biology Y

Philip Hubbard (Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA, USA)
108 2024 Art & Biology Y

Kristen M. Browne (5601 Fishers Ln, Rockville, MD 20852)
109 2024 Art & Biology Y

Tyler Ard, Arthur W. Toga (University of Southern California, Los Angeles, USA)
110 2024 Art & Biology Y

Marco Garbelli (URPP Adaptive Brain Circuits in Development and Learning (AdaBD), University of Zurich, Winterthurerstr. 190 8057 ZÃŒrich)
111 2024 Art & Biology Y

Sneha Tripathi (Indian Institute of Science Education and Research, Pune, India)
112 2024 Art & Biology Y

Harmony Wong (Rochester Institute of Technology, Rochester, New York, United States)
113 2024 Art & Biology Y

Leonora MartÃnez-Nuñez (UMass Chan Medical School. Biochemistry and Molecular Biotechnology Department. Worcester, MA. )
114 2024 Art & Biology Y

Mol Mir, Stephanie Nowotarski, Alejandro Sánchez Alvarado (Stowers Institute for Medical Research, 1000 E 50th St, Kansas City, Missouri, United States)
115 2024 Art & Biology Y

Asim Niyaz, Emel Ergene (Eskisehir Technical University, Turkiye)
116 2024 Art & Biology Y

Jim Stanis, Mike Bienkowski (2025 Zonal Ave. Los Angeles, CA 90033)
117 2024 Art & Biology Y


Alex Ritter (Altos Labs)
119 2024 Art & Biology Y


Hana Pokojná (Masaryk University, Brno, Czech Republic)
121 2023 Genomes A

Joshua Stevenson-Hoare (Cardiff University, Cardiff, UK)
122 2023 Genomes A

Guillermo Marin, Sol Bucalo, Jeronimo Calderon, Alba Jene , Alfonso Valencia, Fernando Cucchietti (Plaça Eusebi GÌell, 1-3, 08034, Barcelona, Spain)
123 2023 Other A

Andrew Waterhouse, Janani Durairaj, Torsten Schwede, Joana Pereira (SIB, University of Basel, Switzerland)
124 2023 Proteins A

Ãloi Durant, François Sabot, Matthieu Conte, Mathieu Rouard ((IRD, Montpellier, France; Syngenta Seeds SAS, Saint-Sauveur, France; Bioversity International, Montpellier, France; South Green Bioinformatics Platform, Montpellier, France))
125 2023 Genomes A

Tyler Ard, Michael S. Bienkowski, James Stanis, Caroline OâDriscoll, Sook-Lei Liew, Arthur W. Toga (University of Southern California, Los Angeles, USA)
126 2023 Genomes A

Nadine Tuechler, Muzamil M Khan, Martin Garrido-Rodriguez, Mira Lea Burtscher, Denes Turei, Rafael Kramann, Susanne Delecluse, Christoph A. Merten, Julio Saez-Rodriguez, Rainer Pepperkok (EMBL Heidelberg, MeyerhofstraÃe 1, 69117 Heidelberg)
127 2023 Cells A

Jakub VaÅ¡ÃÄek, Dafni Skiadopoulou, Ksenia G. Kuznetsova, Stefan Johansson1, PÃ¥l R. NjÞlstad, Stefan Bruckner, Lukas KÀll, Marc Vaudel (Klinisk institutt 2, Universitetet i Bergen, Laboratoriebygget (administrasjon/ekspedisjon i 8 etasje), Haukeland Universitetssykehus, Jonas Lies veg 87, 5021 Bergen, Norway)
128 2023 Proteins A

Rachel Bellone, Pierre Lê-Bury,Christophe Becavin , Catherine Dauga, Olivier Dussurget, Javier Pizarro-Cerda, Pierre Lechat (Institut Pasteur, 28 rue du Docteur Roux 75015 Paris)
129 2023 Genomes A


Ayelen Valko (Heidelberg, Germany)
131 2023 Other A

Mol Mir, Steph Nowotarski, Alejandro Sánchez Alvarado (Stowers Institute for Medical Research, Kansas City, MO, USA)
132 2023 Cells B

Neli Fonseca, Andrii Iudin, Sriram Somasundharam, Zhe Wang, Ryan Pie, Jack Turner, Amudha Duraisamy, Sanja Abbott, Ardan Patwardhan, Gerard Kleywegt (EMBL-EBI, Wellcome Genome Campus, Hinxton, Cambridge, CB10 1SD, UK.)
133 2023 Proteins B

Giulia Ghisleni, Sara Fumagalli, Antonia Bruno, Maurizio Casiraghi (University of Milano-Bicocca, Department of Biotechnology and Biosciences - Piazza dell'Ateneo Nuovo, 1 - 20126, Milan, Italy)
134 2023 Genomes B

Marzia Munafo (EMBL (European Molecular Biology Laboratory) â Epigenetics and Neurobiology Unit, Via Ramarini 32, 00015, Monterotondo (Rome), Italy)
135 2023 Cells B

Denis Bienroth, Natalie Charitakis, Sabrina Jaeger-Honz, Dimitar Garkov, David A.Elliott, Enzo R. Porrello, Karsten Klein, Hieu T. Nim, Falk Schreiber, and Mirana Ramialison (reNEW Novo Nordisk Foundation Center for Stem Cell Medicine & Stem Cell Biology, Murdoch Childrenâs Research Institute, Parkville, Melbourne, VIC, Australia.)
136 2023 Genomes B

Vivek Kumar Srivastav, Angela Kranz and and Björn Usadel (Forschungszentrum JÃŒlich, Wilhelm-Johnen-StraÃe, 52428 JÃŒlich, Germany)
137 2023 Other B

Daniel Wendisch, Oliver Dietrich, Tommaso Mari, Saskia von Stillfried, et al. (see https://doi.org/10.1016/j.cell.2021.11.033)
138 2023 Cells B

Dannielle McCarthy, PhD, and Lucy Obus (https://chanzuckerberg.com/science/programs-resources/imaging/)
139 2023 Cells B

Javier S. Utgés, Stuart A. MacGowan, Callum M. Ives, Geoffrey J. Barton (Division of Computational Biology, School of Life Sciences, University of Dundee, Dundee, UK)
140 2023 Proteins B

Paula Emilia Reimann, Zehra Sude Altunok, Benito Campos, Lars RÞnn Olsen (University of MÌnster, MÌnster, Germany; VAVisual Abstract GmbH, Mannheim, Germany; Charité UniversitÀtsmedizin Berlin, Berlin, Germany; Department of Health Technology, Kongens Lyngby, Denmark)
141 2023 Other B

MichaÅ Åwirski, Hakon Tjeldnes, Joanna Kufel, Eivind Valen (1 Institute of Genetics and Biotechnology, University of Warsaw, Warsaw, Poland,2 Computational Biology Unit, Department of Informatics, University of Bergen, Bergen, Norway)
142 2023 Genomes B

Ãtienne Fafard-Couture, Pierre-Ãtienne Jacques & Michelle S. Scott (Université de Sherbrooke, Sherbrooke, Canada)
143 2023 Genomes C

Kari Lavikka, Jaana Oikkonen, Alexandra Lahtinen, Yilin Li, Sampsa Hautaniemi (University of Helsinki, Helsinki, Finland)
144 2023 Genomes C

Margot Riggi and Janet Iwasa (Department of Biochemistry, University of Utah, Salt Lake City, USA)
145 2023 Genomes C
chemistry education through interactive AR in commodity devices")
Fabio Cortes Rodriguez and Luciano Andres Abriata (School of Life Sciences, Ecole Polytechnique Federale de Lausanne, CH-1015 Lausanne, Switzerland)
146 2023 Proteins C

Chengbo Fu (Aalto University, Espoo, Finland)
147 2023 Genomes C

Sungeun (Kristina) Song, Gabrielle Deschamps-Francoeur, Sherif Abou-Elela, Michelle S. Scott (Université de Sherbrooke, Sherbrooke, Canada)
148 2023 Transcripts C

Fabio Cortes Rodriguez and Luciano Andres Abriata (School of Life Sciences, Ecole Polytechnique Federale de Lausanne, CH-1015 Lausanne, Switzerland)
149 2023 Proteins C

Erik Werner (RNS Berlin UG, c/o betahaus, Rudi-Dutschke-Str. 23, 10969 Berlin)
150 2023 Genomes C

Julia Michalska, Julia Lyudchik,Philipp Velicky, Hana Korinkova, Jake F. Watson, Alban Cenameri, Christoph Sommer, Johann Danzl (Institute of Science and Technology Austria, Am Campus 1, Klosterneuburg, Austria)
151 2023 Cells C

Max Gemmer, Marten Chaillet, Mariska Groellers-Mulderij, Rodrigo Cuevas Arenas, Juliette Fedry, Friedrich Foerster (Structural Biochemistry, Bijvoet Center for Biomolecular Research, Utrecht University, Utrecht, The Netherlands)
152 2023 Proteins C

Blair Lyons, Matthew Akamatsu, Eran Agmon, Allen institute for Cell Science, Graham Johnson (1Allen Institute for Cell Science, Seattle, WA 2Department of Biology, University of Washington, Seattle, WA 3Center for Cell Analysis and Modeling, University of Connecticut, Farmington, CT)
153 2023 Genomes C

Beata Edyta Mierzwa (Ludwig Institute for Cancer Research and University of California San Diego, USA)
154 2023 Other D

Jakub Wojciechowski, Alicja Nowakowska (Wroclaw, wyb. Wyspianskiego 27, Wroclaw University of Science and Technology)
155 2023 Proteins D

Jim Procter, Ben Soares, Geoff Barton (School of Life Sciences, University of Dundee, Scotland, UK.)
156 2023 Proteins D

Matúš TalÄÃk, Filip OpálenÃœ, Tereza Clarence, KatarÃna Furmanová, Jan ByÅ¡ka, Barbora KozlÃková, David KouÅil (Masaryk University, Brno, Czech Republic)
157 2023 Genomes D

Caner Bagci, David Bryant, Banu Cetinkaya and Daniel H. Huson (University of TÃŒbingen, TÃŒbingen, Germany)
158 2023 Genomes D

Aliaksei Chareshneu, Adam Midlik, Alessio Cantara, Crina-Maria Ionescu, Alexander Rose, VladimÃr HorskÃœ, Radka Svobodová, Karel Berka, David Sehnal (National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Brno, 602 00, Czech Republic)
159 2023 Proteins D

Ekaterina Osmekhina, Feng Jianhui, Markus Linder (Allto University, Kemistintie1, Espoo, Finland)
160 2023 Genomes D

Elizaveta Loseva, Aniruddha Mitra, Erwin J.G. Peterman (Vrije Universiteit, Amsterdam, The Netherlands)
161 2023 Cells D


N. Simankov, H. Soyeurt, S. Massart, R. Tahzima. (Gembloux Agro-Bio Tech (ULiÚge), Gembloux, Belgium)
163 2023 Proteins D

Helena Jambor, Christopher Schmied, Michael Nelson, Alex Payne-Dwyer et al. (National Center for Tumor Diseases, University Cancer Center, NCT-UCC UniversitÀtsklinikum Carl Gustav Carus an der Technischen UniversitÀt Dresden FetscherstraÃe 74, 01307 Dresden)
164 2023 Genomes D

Sandeep Kaur, Neblina Sikta, Andrea Schafferhans, Stuart Anderson, Matt Adcock, Nicola Bordin, Mark J. Cowley, David M. Thomas, Mandy L. Ballinger, Seán I. OâDonoghue (Garvan Institute & UNSW, Australia)
165 2023 Genomes D

Tomás Magalhães 1,2,3,4*, AmÃlcar Duarte 1,4, José Alberto Pereira 3,5, Natália Marques 2,4 (1:MED, UAlg Faro, Portugal 2:CEOT, UAlg Faro, Portugal 3:CIMO, IPB, Bragança, Portugal 4:Universidade do Algarve (UAlg), Faro, Portugal 5:Instituto Politécnico de Bragança (IPB), Bragança, Portugal)
166 2023 Proteins A

Berk Turhan 1 , Irene Font Peradejordi 2, Shreya Chandrasekar 2, Selim Kalayci 1, Jeffrey Johnson 1, Mehdi Bouhaddou 3, Zeynep H. GÌmÌŠ1 (1 Department of Genetics and Genomics, Icahn School of Medicine at Mount Sinai, NY; 2 Cornell Tech, Cornell University, NY; 3 Institute for Quantitative and Computational Biosciences, UCLA, CA)
167 2023 Proteins A

Pouneh Pouramini1, Hildur Björg Birnisdóttir2, Jón Már Björnsson2 and Goetz Hensel3 (1Leibniz Institute of Plant Genetics and Crop Plant Research, Seeland, Germany 2ORF Genetics, Reykjavik, Iceland 3Centre for Plant Genome Engineering, Heinrich-Heine-University, Dusseldorf, Germany)
168 2023 Proteins B

Vincenzo Costanzo1, Ludovica Altieri1,2, Matteo Marzi2, Silvia Gasparini2, Carlo Brighi3, Cecilia Mannironi1,2, Patrizia Lavia1,2 ((1)IBPM- Italian National Research Council (CNR), Rome, Italy; (2) Dept of Biology and Biotechnology âCharles Darwinâ, Sapienza University of Rome, Italy; 3) CrestOptics S.p.A. Rome, Italy)
169 2023 Cells B

Pascale Marill, Mo Rahman, Onno Faber, Otavio Good (GeneDrop Inc, San Francisco, USA)
170 2023 Proteins C

Faryal Ashraf , Atia tul Wahab , Muhammad Iqbal Choudhary (Dr . Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Kar ac hi, Karachi 75270, Pakistan)
171 2023 Proteins C

Pedro Machado, Jonathan Moffat, Joshua Lea, Louise Hughes (Oxford Instruments NanoAnalysis, High Wycombe, HP12 3SE, UK)
172 2023 Other C

Andrea Winterbottom, Jamie Allen, Andrey Azov, Jyothish Bhai, Lucy Brooks, Sarah Hunt, Imran Salam, Dan Sheppard, Steve Trevanion, Andres Veidenberg, Robert D. Finn, Andy Yates (European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge, CB10 1SD, United Kingdom)
173 2023 Genomes D
")
Barbora KozlÃková, Marc A. Marti-Renom, Andrea Schafferhans, Nico Scherf, Michelle Scott, Srigokul Upadhyayula, Seán O'Donoghue, James Procter (EMBL, Germany)
174 2023 Art & Biology Y

Ana Catarina Ferreira, MSc (ICVS - Life and Health Sciences Research Institute, University of Minho, Braga, Portugal)
175 2023 Art & Biology Y

Mol Mir (Stowers Institute for Medical Research, Kansas City, MO, USA)
176 2023 Art & Biology Y

Beata Edyta Mierzwa (Ludwig Institute for Cancer Research and University of California San Diego, USA)
177 2023 Art & Biology Y

Jakub Wojciechowski, Alicja Nowakowska (Wroclaw, wyb. Wyspianskiego 27, Wroclaw University of Science and Technology)
178 2023 Art & Biology Y


Guillermo Marin, Sol Bucalo, Jeronimo Calderon, Fernando Cucchietti (Plaça Eusebi GÌell, 1-3, 08034, Barcelona, Spain)
180 2023 Art & Biology Y

Giulia Ghisleni (University of Milano-Bicocca, Department of Biotechnology and Biosciences - Piazza dell'Ateneo Nuovo, 1 - 20126, Milan, Italy)
181 2023 Art & Biology Y

Elizaveta Loseva, Aniruddha Mitra (Vrije Universiteit, Amsterdam, The Netherlands)
182 2023 Art & Biology Y

183 2023 Art & Biology Y

Asim Niyaz, Emel Ergene (Eskisehir Technical University, Turkiye)
184 2023 Art & Biology Y

Marzia Munafo (EMBL (European Molecular Biology Laboratory) â Epigenetics and Neurobiology Unit, Via Ramarini 32, 00015, Monterotondo (Rome), Italy)
185 2023 Art & Biology Y

Dr Fatahiya Kashif (Federal Medical College, Hanna Road, G-8/4, Islamabad, Pakistan)
186 2023 Art & Biology Y

Ekaterina Osmekhina (Allto University, Kemistintie1, Espoo, Finland)
187 2023 Art & Biology Y

Ayelen Valko (Independent artist)
188 2023 Art & Biology Y

Graham Johnson & Thomas Brown, with: Alison Beckett, Ian Prior, Fernanda Cisneros-Soberanis, and William Earnshaw (Seattle, WA, USA)
189 2023 Art & Biology Y


Sophie den Hartog (Living Systems Institute, University of Exeter, UK)
191 2023 Art & Biology Y

Helena Jambor, Christopher Schmied, Michael Nelson, Alex Payne-Dwyer et al. (National Center for Tumor Diseases, University Cancer Center, NCT-UCC UniversitÀtsklinikum Carl Gustav Carus an der Technischen UniversitÀt Dresden FetscherstraÃe 74, 01307 Dresden)
192 2023 Art & Biology Y


Wang J, Youkharibache P, Marchler-Bauer A, Lanczycki C, Zhang D, Lu S, Madej T, Marchler GH, Cheng T, Chong LC, Zhao S, Yang K, Lin J, Cheng Z, Dunn R, Malkaram SA, Tai C-H, Enoma D, Busby B, etc. (National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, 20894, USA)
194 2022 Genomes A

Jim Stanis, Danny J.J. Wang, Xingfeng Shao (2025 Zonal Ave, Los Angeles, CA, 90042)
195 2022 Organisms A

Tyler Ard, Michael S. Bienkowski, Lirong Yan, Sook-Lei Liew, Arthur W. Toga (USC Mark and Mary Stevens Neuroimaging and Informatics Institute, Los Angeles, USA)
196 2022 Other A

Cristoffer Sevilla, Lisa Matthews, Eliot Ragueneau, Chuqiao Gong, Robin Haw, Guanming Wu, Lincoln Stein, Peter DâEustachio and Henning Hermjakob (Hinxton, Cambridge, CB10 1SD, UK)
197 2022 Genomes A

Yordan Hodzhev1, Borislava Tsafarova1, Vladimir Tolchkov1, Nikolay Yanev2, Vania Youroukova2, Silvia Ivanova2, Dimitar Kostadinov2, Stefan Panaiotov1 (1National Center of Infectious and Parasitic Diseases, Sofia, Bulgaria 2Department of Pulmonary Diseases, Medical University, Sofia, Bulgaria)
198 2022 Organisms A

Alberto I. Roca (ProfileGrid.org, Irvine, CA, USA)
199 2022 Genomes A

Neha Goyal, Yahiya Hussain, Gianna Yang, Daniel Haehn (University of Massachusetts-Boston, Boston, US)
200 2022 Other A

Alex Reynolds, Jacob Quon, Cameron Trader, Sabrina Tsai, Ian Johnson, Wouter Meuleman (Altius Institute, 2211 Elliot Avenue, Seattle WA 98121, U.S.A.)
201 2022 Genomes B

Chris Armit (BGI Hong Kong, Hong Kong)
202 2022 Organisms B

Beata Edyta Mierzwa (Ludwig Institute for Cancer Research, and University of California San Diego, USA)
203 2022 Other B

Eliot Ragueneau, Noemi Del-Toro, Anjali Shrivastava, Birgit Meldal, Pablo Porras, Kalpana Panneerselvam, Henning Hermjakob (EMBL-EBI, Wellcome Genome Campus, Hinxton, Cambridgeshire, CB10 1SD, UK. Tel: 44 (0)1223 49 44 44)
204 2022 Genomes B

Stella Tommasi, Tevfik H. Kitapci, Hannah Blumenfeld, and Ahmad Besaratinia (Department of Population and Public Health Sciences, Keck School of Medicine, University of Southern California, Los Angeles, CA)
205 2022 Transcripts B

Charlotte Godard, Corentin Guérinot, Valentin Marcon, Mohamed El Beheiry, Jean-Baptiste Masson (Institut Pasteur, 25-28 rue du Dr Roux, 75015 Paris, FRANCE)
206 2022 Other C

Carla Molins-Pitarch (Elisava, Barcelona School of Design and Engineering (UVic-UCC), Barcelona, Spain)
207 2022 Genomes C

Badri Adhikari (University of Missouri-St. Louis, USA)
208 2022 Proteins C

Nowlan Freese, Karthik Raveendran, Philip Badzuh, Omkar Marne and Ann E. Loraine (UNC Charlotte, Charlotte, NC, USA)
209 2022 Genomes C

Robin Haw, Colin Diesh, Caroline Bridge, Rob Buels, Garrett Stevens, Peter Xie, Teresa Martinez, Elliot Hershberg, Junjun Zhang, Shihab Dider, Scott Cain, Lincoln Stein, Ian Holmes (Ontario Institute for Cancer Research, Toronto, Canada; University of California, Berkeley, USA)
210 2022 Genomes C

Shadi Fuladi, Sarah McGuinness, and Fatemeh Khalili-Araghi (University of Illinois at Chicago)
211 2022 Proteins C

Simon Heumos, Andrea Guarracino, Sven Nahnsen, Erik Garrison (QBiC - University of TÃŒbingen, TÃŒbingen, Germany; Human Technopole, Milan, Italy; QBiC - University of TÃŒbingen, TÃŒbingen, Germany; University of Tennessee Health Science Center, TN, USA)
212 2022 Genomes D

Mol Mir, Stephanie Nowotarski, Alejandro Sánchez Alvarado (1000 East 50th St. Kansas City, MO, USA. )
213 2022 Cells D

Nicolas Fernandez, Justin He, Timothy Wiggin, George Emanuel, Jiang He (61 Moulton St, Cambridge, MA 02138)
214 2022 Transcripts D

Nowlan Freese, Karthik Raveendran, Ann E. Loraine (University of North Carolina at Charlotte,9201 University City Blvd, Charlotte, NC 28223)
215 2022 Genomes D

Martina Maritan, Ludovic Autin, Jonathan Karr, Markus W. Covert, Arthur J. Olson, David S. Goodsell (Scripps Research, San Diego, USA; Icahn School of Medicine at Mount Sinai, New York, USA; Stanford University, Stanford, USA)
216 2022 Cells D

Andrea Schafferhans, Sandeep Kaur, Neblina Sikta, Sean OâÂÂDonoghue (HSWT, Freising, Germany; CSIRO, Sydney, Australia; TUM, Munich, Germany)
217 2022 Genomes D

Sandeep Kaur, Neblina Sikta, Andrea Schafferhans, Nicola Bordin, Mark J. Cowley, David M. Thomas, Mandy L. Ballinger, Seán I. OâDonoghue (UNSW)
218 2022 Proteins D
")
Neblina Sikta, Stuart Anderson, Christian Stolte, Sandeep Kaur, Bosco K. Ho, Nicola Bordin, Matt Adcock, Andrea Schafferhans, Seán I. OâÂÂDonoghue (Garvan Institute of Medical Research)
219 2022 Proteins D

Seán O'Donoghue, James Procter, Björn Sommer, Beata Edyta Mierzwa (Bridge Institute, USC, USA)
220 2022 Art & Biology Y

Mol Mir (1000 East 50th St. Kansas City, MO, USA. )
221 2022 Art & Biology Y

Maja Divjak, PhD (The Peter MacCallum Cancer Centre, Melbourne, Australia)
222 2022 Art & Biology Y

Beata E. Mierzwa & David S. Goodsell (Ludwig Institute for Cancer Research, USA; University of California San Diego, USA; The Scripps Research Institute, USA; Rutgers, the State University of New Jersey, USA)
223 2022 Art & Biology Y


Martina Maritan (Scripps Research, San Diego, USA)
225 2022 Art & Biology Y


Jim Stanis, Ryan Cabeen (2025 Zonal Ave, Los Angeles, CA, 90042)
227 2022 Art & Biology Y

Demian Keihsler (Research Institute of Molecular Pathology (IMP), Campus-Vienna-Biocenter 1, 1030 Vienna, Austria )
228 2022 Art & Biology Y

Christina Schmidt (CECAD Research Center, Faculty of Medicine, University Hospital Cologne, Joseph-Stelzmann-Str. 26, 50931 Cologne, Germany)
229 2022 Art & Biology Y

Aliaksei Chareshneu (1), Purbaj Pant (1), Ravi José Tristão Ramos (1), David Sehnal (1), TuÄrul Gökbel (2), Crina-Maria Ionescu (1), and Jaroslav KoÄa (1) (1)CEITEC, National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Brno, Czech Republic;2)Department of Molecular Biology &Genetics,Izmir Institute of Technology,Izmir,Turkey)
230 2021 Proteins A

Afaf Saaidi, Alain Laederach, Christine E. Heitsch (School of Mathematics, Georgia Institute of Technology)
231 2021 Transcripts A

Martin Wasser, Joo Guan Yeo, Pavanish Kumar, Thaschawee Arkachaisri,Su Li Poh, Jing Yao Leong, Kee Thai Yeo, Salvatore Albani (Translational Immunology Institute, SingHealth DukeNUS Academic Medical Centre, Singapore)
232 2021 Genomes A

Sona Charles and Jeyakumar Natarajan (Bharathiar University)
233 2021 Other A

Kerstin Schmid*, Andreas Knote*, Alexander MÃŒck*, Philipp Ockermann, Keram Pfeiffer, Sebastian von Mammen, Sabine C. Fischer; (*contributed equally) (University of WÃŒrzburg)
234 2021 Other A

Laura Morel, Peter W. Harrison, Guillaume Devailly (GenPhySE, Université de Toulouse, INRAE, ENVT, 31326, Castanet Tolosan, France & EMBL-EBI, Hinxton, United Kindgom)
235 2021 Genomes A
 for fast, inexpensive epidemiological investigations")
Matteo Perini 1, Gherard Batisti Biffignandi 2, Stella Papaleo 1, Alessandro Alvaro 1, Francesco Comandatore 1 (Pediatric Clinical Research Center âRomeo and Enrica Invernizziâ, Università Di Milano, Milan, Italy; University of Pavia, Pavia, Italia)
236 2021 Genomes A

Daniel Krentzel, Romain Laine, Matt Russell, Marie-Charlotte Domart, Ricardo Henriques, Lucy Collinson, Martin Jones (The Francis Crick Institute, London, UK; Imperial College London, UK; University College London, London, UK; Instituto Gulbenkian de Ciência, Oeiras, Portugal)
237 2021 Cells A

Kristen Procko (The University of Texas at Austin, Austin, TX, USA)
238 2021 Other A

Ritik Sinha (IIIT Allahabad)
239 2021 Populations A

Irepan Salvador-MartıÌnez 1, Marco Grillo 2,3, Michalis Averof 2,3 and Maximilian J. Telford 1. (1 University College London, UK. 2 Institut de Génomique Fonctionnelle de Lyon, Lyon, France. and 3 Centre National de la Recherche Scientifique (CNRS), France.)
240 2021 Cells A

Denise Schrama, Cláudia Raposo de Magalhães, Sofia Engrola, Pedro M. Rodrigues (Universidade do Algarve, Campus de Gambelas, Faro, Portugal)
241 2021 Proteins A

Stephen Taylor (MRC Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Headington)
242 2021 Organisms A

Norman Rzepka (scalable minds GmbH, Potsdam, Germany)
243 2021 Cells A

Leah L. Scherschel, N. Heath Patterson, Jeffrey M. Spraggins, Katy Borner (Indiana University - Bloomington, 107 S Indiana Ave, Bloomington, IN 47405)
244 2021 Other A

Duccio Malinverni, Besian Sejdiu, Madan M. Babu (St. Jude Children's Research Hospital, Memphis TN, USA)
245 2021 Proteins A

Marzia Munafo (EMBL - Epigenetics and Neurobiology Unit, Via E. Ramarini 32, 00015 Rome Italy)
246 2021 Genomes A

Jayashree Vijaya Raghavan, Shruthi KS, Vinod Kumar Dorai, Rebecca Diya Samuel, Priyanka Arunachalam, H.C. Chaluvanarayana, Pavan Belahalli, Kalpana S.R., Siddharth Jhunjhunwala (3rd Floor, BSSE, Biological Sciences Building, Indian Institute of Science, Bengaluru, Karnataka, India - 560012)
247 2021 Populations A

Mark Rozanov. Haim J. Wolfson (Tel Aviv University)
248 2021 Proteins A

Christian Stolte (Cellarity, 100 Tech Square, Cambridge, MA 02140, United States)
249 2021 Cells A

Aygen Ergen, Salih Ofluoglu (Mimar Sinan Fine Arts University, Istanbul, TURKEY)
250 2021 Proteins A

Mariana Ascensão-Ferreira, Nuno L. Barbosa-Morais (Instituto de Medicina Molecular João Lobo Antunes - Faculdade de Medicina da Universidade de Lisboa)
251 2021 Transcripts A

Nuno Saraiva-Agostinho, Bernardo P de Almeida, Nuno L Barbosa-Morais (Instituto de Medicina Molecular João Lobo Antunes, Faculdade de Medicina da Universidade de Lisboa, Lisboa, Portugal; Research Institute of Molecular Pathology, Vienna BioCenter, Vienna, Austria)
252 2021 Transcripts A
")
Neblina Sikta (384 Victoria Street)
253 2021 Proteins A

Martina Paganin, Fabio Lauria, Gabriella Viero (via sommarive 18, Trento)
254 2021 Transcripts B

Deborah F. Nacer, Johan Vallon-Christersson, Hans Ehrencrona, Anders Kvist, Ã
ke Borg, Johan Staaf (Oncology, Clinical Sciences Lund, Lund University; Genetics and Pathology, Laboratory Medicine, Region Skåne; Clinical Genetics, Laboratory Medicine, Lund University; all are Lund, Sweden)
255 2021 Populations B

Daisuke Inoue (Build.3, Shiobaru 4-9-1, Minami-ku, Fukuoka, Japan)
256 2021 Proteins B

Kimberly Meechan, Constantin Pape, Valentyna Zinchenko, Martin Schorb, Hernando M. Vergara, Detlev Arendt, Anna Kreshuk, Yannick Schwab, Christian Tischer (EMBL, Heidelberg, Germany; Heidelberg Collaboratory for Image Processing, Ruprecht Karls Universitat Heidelberg, Germany; Sainsbury Wellcome Centre for Neural Circuits and Behaviour, London, UK)
257 2021 Organisms B

Lina M. Gallego-Paez, Jan Mauer (BioMed X Institute, Im Neuenheimer Feld 583, 69120 Heidelberg, Germany)
258 2021 Transcripts B

Masaki Yamabe1, So Nakagawa2, and Akira Wakita1 (1:Graduate School of Media and Governance, Keio University, Japan ; 2:Department of Molecular Life Science, Tokai University School of Medicine, Japan)
259 2021 Genomes B

David Sehnal, Alexander S. Rose, Sebastian Bittrich, Mandar Deshpande, Radka Svobodová, Sameer Velankar, Stephen K. Burley, Jaroslav KoÄa (CEITEC - Central European Institute of Technology, Masaryk University, Brno, 625 00, Czech Republic)
260 2021 Proteins B

Julia Schessner, Ian Wood, Paul Dupree, Georg Borner (Max-Planck Institute of Biochemistry, Martinsried, Germany; University of Cambridge, Cambridge, UK)
261 2021 Proteins B

Bruno Iochins Grisci, Marcio Dorn (Structural Bioinformatics and Computational Biology Lab - Institute of Informatics - Federal University of Rio Grande do Sul, Porto Alegre, Brazil)
262 2021 Cells B

Meta Heidenreich, Saurabh Mathur, Emmanuel D. Levy (Weizmann Institute of Science, Rehovot, Israel)
263 2021 Cells B

Beata Edyta Mierzwa (Ludwig Institute for Cancer Research and University of California San Diego, La Jolla, USA)
264 2021 Other B

Ela Gralinska (Ihnestr. 63)
265 2021 Genomes B

Theresa Harbig, Kay Nieselt (Institute for Bioinformatics and Medical Informatics, University of TÃŒbingen, Germany)
266 2021 Other B

Sumit Bala, Ambarnil Ghosh, Subhra Pradhan (Independant Researcher, Bangalore, India; University College Dublin, Dublin, Ireland; Independant Researcher, Dublin, Ireland)
267 2021 Proteins B

Maria Nikoghosyan, Maria Schmidt, Kristina Margaryan, Henry Loeffler-Wirth, Arsen Arakelyan,Hans Binder (Institute of Molecular Biology NAS RA; Russian-Armenian University; Armenian Bioinformatics Institute; Yerevan State University; Yerevan, Armenia; University of Leipzig, Leipzig, Germany)
268 2021 Genomes B

Jannes Peeters, Jan Aerts (Hasselt University, Hasselt, Belgium)
269 2021 Genomes B

Erik Werner (RNS Berlin, Germany)
270 2021 Other B

Alessia Buratin, Enrico Gaffo, Anna Dal Molin, Stefania Bortoluzzi (Department of Molecular Medicine, University of Padova, Padova, Italy)
271 2021 Transcripts B

Valentin BEAUVAIS, Kévin MOREAU, Aurélia LE DANTEC, A. Rachid RAHMOUNI (CNRS CBM avenue de la recherche scientifique 45071 Orléans Cedex 2)
272 2021 Transcripts B

Alen Adiyev, Dmitry Molotkov, Hiroki Asari (European Molecular Biology Laboratory, Epigenetics and Neurobiology Unit, Rome, Italy)
273 2021 Other B

Andrea Schafferhans, Sandeep Kaur, Neblina Sikta, Christian Dallago, Michael Bernhofer, Burkhard Rost, Sean OâDonoghue (HSWT, Freising, Germany; CSIRO, Sydney, Australia; TUM, Munich, Germany)
274 2021 Proteins B

Chen Hong, Robin Thiele, Lars Feuerbach ( German Cancer Research Center (DKFZ), Heidelberg, Germany)
275 2021 Genomes B

Mol Mir, Stephanie Nowotarski, Melainia McClain, Alejandro Sanchez Alvarado (Stowers Institute for Medical Research, Kansas City MO, USA; Howard Hughes Medical Institute, Chevy Chase MD, USA)
276 2021 Cells B

Adam Pecina, Laura Riccardi, Paolo Scrimin, Fabrizio Mancin and Marco De Vivo (Istituto Italiano di Tecnologia, Via Morego 30, Genova, Italy and Università di Padova, Padova, Italy)
277 2021 Proteins B

Juan Carlos Gonzalez-Sanchez, Mustafa F.R. Ibrahim, Ivo C. Leist, Robert B. Russell (BioQuant, Biochemistry Center (BZH), Heidelberg University, 69120 Heidelberg, Germany)
278 2021 Genomes B

Martina Maritan, Ludovic Autin, Jonathan Karr, Markus Covert, Arthur Olson, David Goodsell (Scripps Research, San Diego, USA; Icahn School of Medicine at Mount Sinai, New York, USA; Stanford University, Stanford, USA)
279 2021 Cells B

Wei Xiong, Lidija Berke, Klaas Bouwmeester, Frank F.M. Becker, Richard Michelmore, Sander Peters, Rob van Treuren, Marieke Jeuken, M. Eric Schranz (Wageningen University and Research, Wageningen, The Netherlands)
280 2021 Genomes C

Dr. Victor Padilla-Sanchez (Washington Metropolitan University)
281 2021 Proteins C

Fabio Marchianò, Annie Yim, Adrien Bonnard, Nuno Miguel Luis, Benoit Giannesini, Alice Carrier, Frank Schnorrer, Bianca Habermann (Aix-Marseille University, CNRS, IBDM UMR 7288)
282 2021 Transcripts C

Hieu T. Nim, Mirana Ramialison (Australian Regenerative Medicine Institute, Monash University, Wellington Road, Clayton, Victoria, Australia; Murdoch Childrenâs Research Institute, Flemington Road, Parkville, Victoria, Australia)
283 2021 Genomes C

Matteo Perino, Per Haberkant, Frank Stein, Raquel Marco-Ferreres, Kathleen-Una Towns, Mikhail Savitski, Eileen E. M. Furlong (EMBL, Heidelberg, Germany)
284 2021 Organisms C

Martina Fröschl (Science Visualization Lab of the University of Applied Arts Vienna, Vienna, Austria)
285 2021 Proteins C

Peter Ilgen, Till Stephan, Stefan Jakobs (MPI for biophysical Chemistry, Am Fassberg 11, 37077 Göttingen)
286 2021 Cells C

Ãloi Durant, François Sabot, Matthieu Conte, Mathieu Rouard (Univ Montpellier - IRD, Montpellier, France; Syngenta Seeds SAS, Saint-Sauveur, France; Bioversity International, Montpellier, France; IFB - South Green Bioinformatics Platform, Montpellier, France)
287 2021 Genomes C

Marlet Morales-Franco, Poonam Bheda, Johannes Becker, Sebastian Maerkl, Carsten Marr, Robert Schneider (Institute of Functional Epigenetics, Helmholtz Zentrum MÃŒnchen, Neuherberg, Germany)
288 2021 Populations C

Yumi L. Briones, Nina Rosario L. Rojas, Fabian Antonio M. Dayrit, Armanda Jerome H. de Jesus Jr., Alexander T. Young (Ateneo de Manila University, Quezon City, Philippines; Institute of Environmental Science and Meteorology, Quezon City, Philippines)
289 2021 Cells C

Ekaterina Osmekhina (Aalto University, Espoo, Finland)
290 2021 Other C

Guoye Guan, Kai Kang, Ming-Kin Wong, Vincy Wing Sze Ho, Zhongying Zhao, Chao Tang (Peking University)
291 2021 Genomes C

Ambra Natalini, Sonia Simonetti, Francesca Di Rosa (ISTITUTO DI BIOLOGIA E PATOLOGIA MOLECOLARI, CONSIGLIO NAZIONALE delle RICERCHE, viale Regina Elena 291, Roma 00161, ITALY)
292 2021 Genomes C

Sandeep Kaur, Timothy Peters, Pengyi Yang, Laurence Luu, Jenny Vuong, Sean O'Donoghue (BioVis, GIMR and CSE, UNSW)
293 2021 Cells C

Carlos Prieto, Ãngela Villaverde and David Barrios (Bioinformatics Service, Nucleus, University of Salamanca.)
294 2021 Genomes C

P. Le Mercier, J. Bolleman, E. de Castro, A. Auchincloss, E. Gasteiger, E. Boutet, L. Breuza, C. Casals, A. Estreicher, M. Feuermann, D. Lieberherr, C. Rivoire, I. Pedruzzi, N. Redaschi and A. Bridge. (Swiss-Prot group, SIB Swiss Institute of Bioinformatics, Geneva, Switzerland)
295 2021 Cells C

Nowlan Freese, Karthik Raveendran, Chester Dias, Ann E. Loraine (University of North Carolina at Charlotte, Charlotte, NC U.S.A.)
296 2021 Genomes C

SJ Roostee, J Staaf, M Aine (Department of Clinical Sciences Division of Oncology Lund University Medicon Village SE-223 81 Lund Sweden)
297 2021 Other C


Nurul 'Ayn Ahmad Sayuti, Bjorn Sommer (Royal College of Art, Kensington Gore)
299 2021 Genomes C

Agostinetto G., Sandionigi A., Bruno A., Casiraghi M., Pescini D. (University of Milano-Bicocca, Milan, Italy; Quantia Consulting srl, Milan, Italy)
300 2021 Other C

Lance D. Hentges, Martin J. Sergeant, Damien J. Downes, Jim R. Hughes, Stephen Taylor (University of Oxford MRC WIMM Centre for Computational Biology, Oxford, UK)
301 2021 Genomes C

Tatjana Hirschmugl (Freelance Scientific Illustrator)
302 2021 Genomes C

Johannes Waschke, Mario Hlawitschkaââ, Kerim Anlasâ, Vikas Trivedi, Ingo Roeder, Jan Huisken, and Nico Scherf (*) (MPl for Human Cognitive and Brain Sciences, Leipzig, Germany; HTWK Leipzig, Germany; EMBL Barcelona, Spain; EMBL Heidelberg, Germany; TU Dresden, Germany; Morgridge Institute for Research,Madison,USA)
303 2021 Cells D
 model to dissect the phenotype of FH loss")
Christina Schmidt, Ariane Mora, Lorea Valcarcel, Laura Tronci, Efterpi Nikitopoulou, Ming Yang, Sabrina Rossie, Alexander von Kriegsheim, Mikael Boden and Christian Frezza (Box 197 Cambridge Biomedical Campus, Hutchsion/MRC Research Centre)
304 2021 Cells D

Yordan Hodzhev, Borislava Tsafarova, Vladimir Tolchkov, Stefan Panaiotov (National Center for Infectious and Parasitic Diseases, Sofia, Bulgaria)
305 2021 Genomes D

Ryan Lane (TU Delft, Delft, Netherlands)
306 2021 Cells D


Kari Lavikka, Jaana Oikkonen, Rainer Lehtonen, Johanna Hynninen, Sakari Hietanen, Sampsa Hautaniemi (University of Helsinki, Faculty of Medicine, Helsinki, Finland)
308 2021 Genomes D

Adam Pestana Motala, Christopher Peddie, Anne Weston, Luke Nightingale, Joost de Folter, Amy Strange, Helen Spiers, Lucy Collinson and Martin Jones (The Francis Crick Institute, 1 Midland Rd, Somers Town, London, NW1 1AT)
309 2021 Cells D

Patricia Bondia, Cristina Flors and Joaquim Torra (Madrid Institute for Advanced Studies in Nanoscience (IMDEA nanoscience), Ciudad Universitaria de Cantoblanco. Faraday 9, 28049, Madrid, Spain)
310 2021 Cells D

Tyler F Beck, Mark Williams, Dac-Trung Nguyen, Biomedical Data Translator Consortium, Noel Southall, Christine Colvis (Bethesda, MD)
311 2021 Genomes D

Neringa Daugelaviciene, Ausra Sasnauskiene, Urte Neniskyte, Daiva Dabkeviciene (SaulÄtekio av. 7, LT- 10257 Vilnius, Lithuania)
312 2021 Cells D

S. Indino, E. Rosi, F.W. Baruffaldi Preis, F. Prignano, E. Donetti (via Luigi Mangiagalli, 31, Milan)
313 2021 Cells D

PDB Component Library Collaboration, Presented by Mandar Deshpande (EMBL-EBI)
314 2021 Proteins D

J. Hornby*, B. van Rossum**, S. Thapa*, R. Metzler* and G. McGill*** (University of Potsdam Faculty of Science Karl-Liebknecht-Str. 24-25 14476 Potsdam OT Golm)
315 2021 Proteins D

Eva-Maria Weiss, Aneta Petruskova, Anna Fejtova (Department Psychiatry and Psychotherapy, University Hospital Erlangen,Erlangen, Germany;Charles University, Third Faculty of Medicine, Prague, CZ;National Institute of Mental Health, Klecany, CZ)
316 2021 Genomes D

Nowlan Freese, Karthik Raveendran, Rachel Weidenhammer, Philip Badzuh, Omkar Marne, Chirag Shetty, Irvin Naylor, Chester Dias and Ann E. Loraine (University of North Carolina at Charlotte, Charlotte, U.S.A.)
317 2021 Genomes D

Christoph C.H. Langer, Michael Mitter, Daniel W. Gerlich (Dr.-Bohr-Gasse 3)
318 2021 Genomes D

Henning Hermjakob, The Reactome Consortium (EMBL-EBI, Wellcome Genome Campus, Hinxton, Cambridgeshire, UK)
319 2021 Cells D

Nina M. Billows, Jody E. Phelan, Dong Xia, Yonghong Peng, Taane G. Clark, Yu-Mei Chang (nbillows@rvc.ac.uk)
320 2021 Genomes D

Agnieszka StrzaÅka, Marcin Szafran, Dagmara Jakimowicz (Biotechnology Department, University of Wroclaw)
321 2021 Genomes D

Jelmer Bot, Dries Testelmans, Jan Aerts (Hasselt University, Hasselt, Belgium; Department of Respiratory Diseases, Leuven University Hospital, Leuven, Belgium;)
322 2021 Populations D

Roman Baptista, Xia Dong, Laura Toni (4 Royal College St, London NW1 0TU)
323 2021 Cells D

Heba Sailem (Institute of Biomedical Engineering, University of Oxford)
324 2021 Cells D

QBI Coronavirus Research Group, Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, Polacco BJ, Melnyk JE, Ulferts S, Kaake RM, Batra J, et al. (QBI COVID-19 Research Group, San Francisco, USA; Quantitative Biosciences Institute, San Francisco, USA; University of California, San Francisco, USA; EMBL-EBI, Cambridge, UK.)
325 2021 Proteins D

Gael McGill (138 Fuller Street, apt #1)
326 2021 Proteins D
")
Sean I. O'Donoghue, James Procter, Bjorn Sommer (EMBL Heidelberg, Germany)
327 2021 Art & Biology Y

Emilyn Frohn (University of Illinois at Chicago, Chicago, IL)
328 2021 Art & Biology Y


Deborah Figueiredo Nacer de Oliveira (Div of Oncology, Dept of Clinical Sciences Lund, Lund University, Medicon Village, Lund, Sweden)
330 2021 Art & Biology Y


Concept by V. Zanotelli, B. Bodenmiller and colleagues, illustrated by Christine Shan (SciStories LLC)
332 2021 Art & Biology Y

Dr. Victor Padilla-Sanchez (Washington Metropolitan University)
333 2021 Art & Biology Y


Fabio Marchianò (Aix-Marseille University, CNRS, IBDM UMR 7288)
335 2021 Art & Biology Y

Marzia Munafo (EMBL - Epigenetics and Neurobiology Unit, Via E. Ramarini 32, 00015 Rome Italy)
336 2021 Art & Biology Y

Johannes Waschke, Mario Hlawitschkaââ, Kerim Anlasâ, Vikas Trivedi, Ingo Roeder, Jan Huisken, and Nico Scherf (*) (MPl for Human Cognitive and Brain Sciences, Leipzig, Germany; HTWK Leipzig, Germany; EMBL Barcelona, Spain; EMBL Heidelberg, Germany; TU Dresden, Germany; Morgridge Institute for Research,Madison,USA)
337 2021 Art & Biology Y

Sigrid Knemeyer (SciStories LLC) (SciStories LLC)
338 2021 Art & Biology Y

Christina Schmidt (Box 197, Cambridge Biomedical Campus)
339 2021 Art & Biology Y

Meta Heidenreich (Weizmann Institute of Science, Rehovot, Israel)
340 2021 Art & Biology Y


Iliana Oakes (School of Creative Industries, University of Newcastle, Australia)
342 2021 Art & Biology Y

Martina Fröschl (Science Visualization Lab of the University of Applied Arts Vienna, Vienna, Austria)
343 2021 Art & Biology Y

Patricia Bondia, Cristina Flors and Joaquim Torra (Madrid Institute for Advanced Studies in Nanoscience (IMDEA nanoscience), Ciudad Universitaria de Cantoblanco. Faraday 9, 28049, Madrid, Spain)
344 2021 Art & Biology Y

Peter Ilgen, Till Stephan, Stefan Jakobs (MPI for biophysical Chemistry, Am Fassberg 11, 37077 Göttingen)
345 2021 Art & Biology Y

Bruno Iochins Grisci, Marcio Dorn (Structural Bioinformatics and Computational Biology Lab - Institute of Informatics - Federal University of Rio Grande do Sul, Porto Alegre, Brazil)
346 2021 Art & Biology Y

Gianluca Tomasello, Martina Maritan (3D Protein Imaging, Varese, Italy; Scripps Research, San Diego, USA)
347 2021 Art & Biology Y

KarolÃna KryÅ¡tofová (Masaryk University, Faculty of Science, Brno, Czech Republic)
348 2021 Art & Biology Y

Beata Edyta Mierzwa (Ludwig Institute for Cancer Research and University of California San Diego, La Jolla, USA)
349 2021 Art & Biology Y

Ekaterina Osmekhina (Aalto University, Espoo, Finland)
350 2021 Art & Biology Y

Heiko Stark (Friedrich-Schiller-University Jena, Jena, Germany)
351 2021 Art & Biology Y

Eva Klose, Gideon Bergheim (Eva Klose - Design, Heidelberg and IPMB, Heidelberg)
352 2021 Art & Biology Y

Elisabed Zaldastanishvili (G. Eliava Institute of Bacteriophages, Microbiology and Virology (Tbilisi, Georgia))
353 2021 Art & Biology Y

Leonora MartÃnez-Nunez, Mary Munson (UMASS medical school. Worcester, Massachusetts. )
354 2021 Art & Biology Y


Yordan Hodzhev, Borislava Tsafarova, Vladimir Tolchkov, Stefan Panaiotov (National Center for Infectious and Parasitic Diseases, Sofia, Bulgaria)
356 2021 Art & Biology Y

Shahab Mirshahvaladi (Macquarie University, Sydney, Australia)
357 2021 Art & Biology Y

Jayashree Vijaya Raghavan, Shruthi KS, Vinod Kumar Dorai, Rebecca Diya Samuel, Priyanka Arunachalam, H.C. Chaluvanarayana, Pavan Belahalli, Kalpana S.R., Siddharth Jhunjhunwala (3rd Floor, BSSE, Biological Sciences Building, Indian Institute of Science, Bengaluru, Karnataka, India - 560012)
358 2021 Art & Biology Y

Gary Oberbrunner (Dark Star Systems, Somerville MA, USA)
359 2021 Art & Biology Y
 helping fight COVID-19")
Mandar Deshpande (EMBL-EBI)
360 2021 Art & Biology Y

Agostinetto Giulia (University of Milano-Bicocca, Milan, Italy)
361 2021 Art & Biology Y


Helena Pinheiro (Instituto de Medicina Molecular, Lisboa, Portugal)
363 2021 Art & Biology Y

Heidrun Gundlach (PGSB - Plant Genome and Systems Biology, Helmholtz Center Munich, Germany)
364 2021 Art & Biology Y

Paraskevi Vlachou (https://www.dkfz.de/en/immundiversitaet/index.php)
365 2021 Art & Biology Y

Heba Sailem (Institute of Biomedical Engineering, University of Oxford)
366 2021 Art & Biology Y

Joel Hornby, Molecular Visuals (University of Potsdam Faculty of Science Karl-Liebknecht-Str. 24-25 14476 Potsdam OT Golm)
367 2021 Art & Biology Y
")

Maya Voichek (Shamir) (Weizmann Institute of Science, Rehovot 7610001, Israel)
369 2019 Cells A

Daigo Okada, Naotoshi Nakamura, Kazuya Setoh, Takahisa Kawaguchi, Maiko Narahara, Koichiro Higasa, Yasuharu Tabara, Fumihiko Matsuda, Ryo Yamada (Center for Genomic Medicine, Graduate School of Medicine Kyoto University Nanbusogo-Kenkyu-To-1, 5F 53 Syogoin-Kawaharacho, Sakyo-ku, Kyoto)
370 2019 Cells A

Aaron Watters (Flatiron Institute, New York City, NY, USA)
371 2019 Transcripts A

Rebecca Kirsch, Ole N. Jensen, Veit SchwÀmmle (Protein Research Group, University of Southern Denmark, Odense)
372 2019 Cells A

Colas Droin, Jakob El Kholtei, Keren Bahar Halpern, Clémence Hurni, Shalev Itzkovitz, Félix Naef (EPFL, Lausanne)
373 2019 Transcripts A

Liis Kolberg, Uku Raudvere, Jaak Vilo, Hedi Peterson (University of Tartu, Tartu, Estonia)
374 2019 Other A

Terezia Horvathova, Vladimir Sustr, Roey Angel, Ulf Bauchinger (Institute of Soil Biology Biology Centre CAS, Ceske Budejovice, Czechia; Institute of Environmental Sciences Jagiellonian University, Krakow, Poland)
375 2019 Organisms A

Konstantinos Sidiropoulos, Chuqiao Gong, Pascual Lorente, Guilherme Viteri, Lincoln Stein, Peter DâEustachio, Guanming Wu, Antonio Fabregat, and Henning Hermjakob (European Bioinformatics Institute (EMBL-EBI), Cambridge, UK)
376 2019 Cells A

Fatima Heinicke, Albert Pla Planas, Siqing Liu, Yafei Xing, Xiangfu Zhong, Simon Rayner (Oslo University Hospital, Kirkenveien 166, 0465 Oslo)
377 2019 Genomes A

Wencke Walter, Claudia Haferlach, Wolfgang Kern, Torsten Haferlach, Manja Meggendorfer (MLL MÌnchner LeukÀmielabor GmbH Max-Lebsche-Platz 31, 81377 Munich)
378 2019 Genomes A

Elizabeth Ing-Simmons, Juan M. Vaquerizas (Max Planck Institute for Molecular Biomedicine, Muenster, Germany)
379 2019 Genomes A


Andreas Knote, David Bernard, Sylvain Cussat-Blanc and Sebastian von Mammen (Games Engineering, Chair for Human-Computer-Interaction, University of WÃŒrzburg, Germany; ITAV, IRIT, CNRS France;)
381 2019 Genomes A

Matthias Blum, Pierre-Etienne Cholley, Marco Antonio Mendoza-Parra, Hinrich Gronemeyer (Department of Functional Genomics and Cancer, Institut de Génétique et de Biologie Moléculaire et Cellulaire, University of Strasbourg, 67404 Illkirch, France)
382 2019 Genomes A

Michael Zager, Gretchen Krenn (Fred Hutch Cancer Center, Seattle, WA, USA)
383 2019 Genomes A

Bikash Ranjan Samal, Alagar S, Ranjit Prasad Bahadur (IIT Kharagpur, West Bengal, India, Pin code - 721302)
384 2019 Other B

Megumi Inoue, W. Matthew Leevy (University of Notre Dame, Notre Dame, IN 46556, USA. )
385 2019 Other B

Roman Hillje, Lucilla Luzi, Pier Giuseppe Pelicci (Istituto Europeo di Oncologia, Milan, Italy)
386 2019 Transcripts B


Martin Graham, Colin Combe, Lars Kolbowski, Juri Rappsilber (Wellcome Centre for Cell Biology, Michael Swann Building, University of Edinburgh & Dept. of Bioanalytics, TU Berlin)
388 2019 Genomes B

David Barrios and Carlos Prieto (University of Salamanca, Spain)
389 2019 Genomes B


Adrien Georges, Thomas Cloatre, Lu Liu, Délia Dupré, Siying Huang, Romain Glandier, Takiy Berrandou, ARCADIA study, Nabila Bouatia-Naji (PARCC/HEGP, Paris, France)
391 2019 Genomes B

Wilhelm Bertrams (Institute for Lung Research, Marburg)
392 2019 Genomes B

Luke Whitehorn (DEMCON | nymus3D, Institutenweg 25, 7521PH Enschede, the Netherlands)
393 2019 Genomes B

Carmen Mata Martin, Yan Ning Zhou, Ding Jun Jin (Transcription Control Section, RNA Biology Laboratory, CCR, National Cancer Institute, NIH, Frederick, MD 21702-1201)
394 2019 Genomes B

Luke Donnelly, Justin Magee, Niall Haslam, Shane Wilson (Ulster University, Magee Campus, Northland Rd, Londonderry BT48 7JL)
395 2019 Other B

Nuno Saraiva-Agostinho, Nuno Barbosa-Morais (Instituto de Medicina Molecular João Lobo Antunes, Universidade de Lisboa, Lisboa, Portugal)
396 2019 Transcripts B

Kashyap Chhatbar, Raphaël Pantier, Timo Quante, Konstantina Skourti-Stathaki, Jim Selfridge, Adrian Bird (Wellcome Centre for Cell Biology, University of Edinburgh, Michael Swann Building, Max Born Crescent, EDINBURGH, EH9 3BF, U. K.)
397 2019 Transcripts B


Bosco Ho, Michael Joss, Andrea Schafferhans, Seán OâDonoghue (Garvan Institute of Medical Research)
399 2019 Proteins B

Alex Diaz-Papkovich, Luke Anderson-Trocme, Simon Gravel (McGill University, Montreal, Canada.)
400 2019 Genomes C

Yiming Cheng, Li Jiang, Susanne Keipert, Shuyue Zhang, Andreas Hauser, Elisabeth Graf, Tim Strom, Matthias Tschoep, Martin Jastroch, and Fabiana Perocchi (Institute for Diabetes and Obesity, Helmholtz Diabetes Center, Helmholtz Zentrum Muenchen and German National Diabetes Center (DZD), 85764 Neuherberg, Germany)
401 2019 Other C

Beata Edyta Mierzwa (Ludwig Institute for Cancer Research and University of California San Diego, La Jolla, USA)
402 2019 Other C

Carlos Prieto and David Barrios (Bioinformatics Service, Nucleus, University of Salamanca.)
403 2019 Genomes C

Jessica Janson, Claudio Babiloni, Roberta Lizio (Department of Physiology and Pharmacology âV. Erspamerâ at Sapienza University of Rome, Piazza Aldo Moro 5, 00185 Roma, Italy )
404 2019 Other C

Chris Armit, Si Zhe Xiao, Scott Edmunds, Laurie Goodman, Peter Li, Mary Ann Tuli, Christopher Ian Hunter (GigaScience, BGI-Hong Kong)
405 2019 Organisms C

Radek Fedr, Ján RemÅ¡Ãk, JiÅà Navrátil, Lucia Binó, Eva Slabáková, Pavel Fabian, Marek Svoboda, and Karel SouÄek (1-Institute of Biophysics of the Czech Academy of Sciences, CZ; 2-Center of Biomolecular and Cellular Engineering, International Clinical Research Center, St. Anne's University Hospital Brno, CZ)
406 2019 Populations C

Neta Varsano1, Xueting Jin2, Tali Dadosh1, Nadav Elad1, Eva Pereiro3, Ana J Perez-Berna3, Fabio Beghi1, Howard S. Kruth2, Leslie Leiserowitz1, and Lia Addadi1 (1Weizmann Institute of Science, Israel. 2National Institutes of Health, , MD,USA 3ALBA Synchrotron Light Source, Spain )
407 2019 Organisms C

Loïc Meunier, Philippe Jacques, Denis Baurain (University of LiÚge, LiÚge, Belgium)
408 2019 Genomes C

Waterhouse, A., Robin, X., Bienert, S., de Beer, T.A.P., Tauriello, G., Studer, G., Bordoli, L., Schwede, T. (Biozentrum, University of Basel, Switzerland and SIB, Swiss Institute of Bioinformatics, Klingelbergstrasse 50â70, CH-4056 Basel, Switzerland)
409 2019 Proteins C

Jyothish Bhai, Sanjay Boddu, Imran Salam, Dan Sheppard, Steve Trevanion, Andrea Winterbottom, Andy Yates, Paul Flicek (European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge, CB10 1SD, United Kingdom)
410 2019 Genomes C

Andreas Harmuth, Mike Bogetofte Barnkob, Lars RÞnn Olsen (Department of Health Technology, Technical University of Denmark)
411 2019 Other C

Jelena Äuklina, Chloe Lee, Evan G. Williams, Tatjana Sajic, Ben Collins, MarÃa RodrÃguez MartÃnez, Ruedi Aebersold (Otto-Stern-Weg 3, ZÃŒrich, Switzerland; Ph.D. Program in Systems Biology, Zurich, Switzerland; IBM Research, RÃŒschlikon, Switzerland; University of Zurich, Switzerland)
412 2019 Genomes C

Karel Drbal (Faculty of Science, Charles University, Prague, Czechia)
413 2019 Cells C

Christian Stolte, Kevin Shi, Sylvestre Gug, Nina Lapchyk, Nathaniel Novod, Avinash Abhyankar, and Toby Bloom (New York Genome Center, New York, USA)
414 2019 Genomes C

Tyler Beck, Mark Williams, Tyler Peryea, Dac-Trung Nguyen, Biomedical Data Translator Consortium, Noel Southall, Christine Clovis (NCATS, Bethesda, MD, USA)
415 2019 Other D

Wiseman, B., Nitharwal, R.G., Fedotovskaya, O., SchÀfer, J., Guo, H., Kuang, Q., Benlekbir, S., Sjöstrand, D., Ãdelroth, P., Rubinstein, J.L., Brzezinski, P. and Högbom, M. (Department of Biochemistry and Biophysics, Stockholm University, Stockholm, Sweden)
416 2019 Genomes D

1Polesel M, 1Schuh CD, 1,2HÀnni D, 2Kirschmann M, 2Mateos JM 2KÀch A, 1Hall AM (1Structural & Functional Imaging Group, Institute of Anatomy, University of Zurich, Switzerland 2Center for Microscopy and Image Analysis, University of Zurich, Switzerland)
417 2019 Organisms D

Jules Kerssemakers (Omics IT & Datamanagement Core Facility (W610), Deutsches Krebsforschungszentrum (DKFZ) Heidelberg)
418 2019 Genomes D

PDBe-KB Consortium, Presented by Mandar Deshpande (EMBL-EBI)
419 2019 Proteins D

Stefano Vianello, Matthias Lutolf (EPFL, Lausanne, Switzerland)
420 2019 Organisms D

Fanny Georgi (Institute of Molecular Life Sciences, University of Zurich, Zurich, Switzerland)
421 2019 Populations D

Frank Siedler, Kristina A. Ganzinger, Petra Schwille (MPI of Biochemistry, Martinsried, Germany; AMOLF, Amsterdam, The Netherlands; MPI of Biochemistry, Martinsried, Germany)
422 2019 Other D

Pascual Lorente (1), Konstantinos Sidiropoulos (1), Guilherme Viteri(1), Lincoln Stein (2,3), Guanming Wu (4), Peter dâEustachio (5), Henning Hermjakob (1,6), Antonio Fabregat (1,7) (1) EMBL-EBI, UK 2) OICR, Canada 3) Dept of Molecular Genetics, University of Toronto, Canada 4) OHSU, USA. 5) NYU Langone Medical Center, USA 6) State Key Lab. of Proteomics, China 7) Open Targets, UK)
423 2019 Genomes D

Yang Ni, Muhammad Hagras, Vassiliki Konstantopoulou, Johannes Mayr, Alexei Stuchebrukhov, David Meierhofer (Max Planck Institute for Molecular Genetics, Berlin; Freie UniversitÀt Berlin, Berlin, Germany; UC Davis, USA; Medical Univeristy of Vienna, Austria; Paracelsus Medical University, Salzburg, Austria)
424 2019 Cells D

Régis Ongaro-Carcy, Mickael Leclercq, Adrien Dessemond, Marie-Pier Scott-Boyer, François Belleau, Olivier Perin, Arnaud Droit (Computational Biology Laboratory, Centre de recherche du CHU de Québec - Université Laval 2705 boul. Laurier, R-4773)
425 2019 Other D

Mehmood Ghaffar, Niklas Biere, Falk Schreiber, Bjorn Sommer (University of Konstanz, Universitaetsstr. 10, 78457 Konstanz, Germany)
426 2019 Genomes D

Christoph C. H. Langer and Daniel W. Gerlich (Institute of Molecular Biotechnology of the Austrian Academy of Sciences (IMBA), Vienna BioCenter (VBC), Vienna, Austria.)
427 2019 Genomes D

Benedetta Frida Baldi, Michael Joss, Kenny Sabir, Christian Stolte, Susan Clark, Sean OâDonoghue (Garvan Institute of Medical Research)
428 2019 Genomes D


Mayank Modi (Turku Centre for Biotechnology, University of Turku & Ã
bo Akademi University, Turku, Finland)
430 2019 Art & Biology Y

Maya Voichek (Shamir) (Weizmann Institute of Science, Rehovot, Israel)
431 2019 Art & Biology Y

Sigrid Knemeyer (19 Fairview Dr, Southborough, MA 01772, USA)
432 2019 Art & Biology Y

Wiseman, B., Nitharwal, R.G., Fedotovskaya, O., SchÀfer, J., Guo, H., Kuang, Q., Benlekbir, S., Sjöstrand, D., Ãdelroth, P., Rubinstein, J.L., Brzezinski, P. and Högbom, M. (Department of Biochemistry and Biophysics, Stockholm University, Stockholm, Sweden)
433 2019 Art & Biology Y

Beata Edyta Mierzwa (Ludwig Institute for Cancer Research and University of California San Diego, La Jolla, USA)
434 2019 Art & Biology Y

Margot Riggi (University of Geneva, 30 quai Ernest Ansermet, 1211 Geneva, Switzerland)
435 2019 Art & Biology Y

Marcello Polesel, PhD candidate (Institute of Anatomy, University of Zurich. Winterthurerstrasse 190 8057 ZÃŒrich, Switzerland)
436 2019 Art & Biology Y


Anna Hupalowska (Broad Institute of MIT and Harvard, Cambridge, MA)
438 2019 Art & Biology Y

Maja Divjak (The Peter MacCallum Cancer Centre, Melbourne, Australia)
439 2019 Art & Biology Y

Fanny Georgi (Institute of Molecular Life Sciences, University of Zurich, Zurich, Switzerland)
440 2019 Art & Biology Y


Agnieszka Kawska, David Prangishvili (Institut Pasteur, Department of Microbiology, 25, rue du Dr. Roux, 75015 Paris, France)
442 2019 Art & Biology Y

Luis Lopes (DEMCON | nymus3D, Institutenweg 25, 7521PH Enschede, the Netherlands)
443 2019 Art & Biology Y

Miguel Motta (DEMCON | nymus3D, Institutenweg 25, 7521PH Enschede, the Netherlands)
444 2019 Art & Biology Y

Monia ZoppÚ - SciVis (CNR, Via Moruzzi 1, Pisa. Italy)
445 2019 Art & Biology Y

Wencke Walter, Claudia Haferlach, Wolfgang Kern, Torsten Haferlach, Manja Meggendorfer (MLL MÌnchner LeukÀmielabor GmbH Max-Lebsche-Platz 31, 81377 Munich)
446 2019 Art & Biology Y

Sébastien Gauvrit (Max Planck Institute for Heart and Lung Research, Bad Nauheim, Germany)
447 2019 Art & Biology Y

Carmen Mata Martin, Yan Ning Zhou, Ding Jun Jin (Transcription Control Section, RNA Biology Laboratory, CCR, National Cancer Institute, NIH, Frederick, MD 21702-1201)
448 2019 Art & Biology Y

Valerie Altounian and Chris Burns, Science/AAAS (1200 New York Ave NW, Washington, DC)
449 2019 Art & Biology Y

Renaud Chabrier (Institut Curie)
450 2019 Art & Biology Y

Tatjana Hirschmugl (I have not been implicated in the research on which the poster/image is based. The original publication is cited at the bottom. )
451 2019 Art & Biology Y

Alex Diaz-Papkovich, Luke Anderson-Trocme, Simon Gravel (McGill University, Montreal, Canada.)
452 2019 Art & Biology Y

Chris Armit, Si Zhe Xiao, Scott Edmunds, Laurie Goodman, Peter Li, Mary Ann Tuli, Christopher Ian Hunter (GigaScience, BGI-Hong Kong)
453 2019 Art & Biology Y


Jessica Hu (University of Copenhagen)
455 2019 Art & Biology Y

Sean O'Donoghue, James Procter, Bang Wong (Broad Institute of MIT & Harvard)
456 2018 Art & Biology Y

Diego Pitta de Araujo1, Steven Wolf1, Andrew Wong1, Sruti Jagannathan1, Lakshmi Ramachandran1, Wei Ting Melanie Lee1, D. DuPuy1, Kyaw Tun1, Jean-Francois Rupprecht1, Michael Sheetz1,2 (1 Mechanobiology Institute, National University of Singapore, Singapore; 2 Dept. of Biological Sciences, Columbia University, USA.)
457 2018 Cells A

Boris Fedorov, Anatoliy Kuznetsov, Victor Joukov and Tatiana Tatusova (National Center for Biotechnology Information, National Institutes of Health Bethesda, MD, 20894 USA)
458 2018 Genomes A

Kyle McClary (The Bridge Institute at USC, 1002 Childs Way, Los Angeles, CA 90089)
459 2018 Other A

John Francis, Raymond Stevens, Fei Sha, Alex McDowell (University of Southern California, Los Angeles, USA)
460 2018 Genomes A

RCSB PDB team at Rutgers and UCSD. Presented by Maria Voigt (RCSB Protein Data Bank | Rutgers, The State University of New Jersey |Center for Integrative Proteomics Research | 174 Frelinghuysen Rd, Piscataway, NJ 08854-8076)
461 2018 Proteins A

Anh Nguyen, Sara Tavana (20 Somerset, Boston, MA, 02111)
462 2018 Other A

Katherine Huang, François Aguet, Kane Hadley, Duyen Nguyen, Jared Nedzel, Kristin Ardlie (Broad Institute of Harvard and MIT, Cambridge, USA)
463 2018 Transcripts A

Jacki Whisenant, Jan Huisken (Morgridge Institute for Research, Madison, United States of America; University of Wisconsin-Madison, Madison, United States of America)
464 2018 Organisms A

Eric Weitz, Jonathan Bistline, Timothy Tickle, Scott Sutherland (Broad Institute of MIT and Harvard, Cambridge, MA, United States of America)
465 2018 Genomes A

Maria Nattestad, Calvin Bao, Michael C. Schatz, Brett Hannigan (DNAnexus, Mountain View, CA, USA; Johns Hopkins, Baltimore, MD, USA)
466 2018 Genomes B

Marwan Abdellah (Geneva 1202, Switzerland )
467 2018 Cells B

Jiaye He, Jan Huisken (330 N Orchard St. Madison, WI, 53715, USA)
468 2018 Other B

Michael Joss, Aaron Thornton, Bosco Ho, Seán O'Donoghue (CSIRO, Sydney Australia)
469 2018 Genomes B

Achyut Sapkota, Kazunori Hamada (National Institute of Technology, Kisarazu College, 2-11-1, Kiyomidai-Higashi, Kisarazu, Chiba 292-0016 Japan)
470 2018 Other B

Lisa Schneider, Peter Sarkies (MRC London Institue of Medical Sciences, London, UK; Imperial College London, London, UK;)
471 2018 Genomes B

Authors: Laura Urbanski, Nathan Leclair, Olga Anczuków*, image: Matt Wimsatt (The Jackson Laboratory for Genomic Medicine, 10 Discovery Drive, Farmington, CT 06032)
472 2018 Transcripts B

Evan Molinelli, Laura Garrison, Frank Sculli (594 Broadway, Suite 1101, New York, NY 10012)
473 2018 Genomes B

Lorena Pantano, Francisco Pantano, Eulalia Marti, Shannan Ho Sui (1 Harvard T.H. Chan School of Public Health, Boston)
474 2018 Transcripts C

Jenny Vuong, Sandeep Kaur, Seán O'Donoghue (Data61, CSIRO, Australia; UNSW, Sydney; Garvan Institute of Medical Research)
475 2018 Cells C

Benoit Bouvrette, Louis Philip; Wang, Xiaofeng; Cody, Neal A.L; Bergalet, Julie; Lefebvre, Fabio Alexis; Bovaird, Samantha; Blanchette, Mathieu; Lécuyer, Éric (Institut de Recherches Clinique de Montréal, Montréal, Canada; Département de Biochimie, Université de Montréal, Montréal, Canada; School of Computer Science, McGill University, Montréal, Canada)
476 2018 Transcripts C

Liis Kolberg, Hedi Peterson (University of Tartu, Tartu, Estonia)
477 2018 Genomes C
")
Melanie B. Brewer, Gurupartap Davis, Nevan Krogan (The HARC Center and Quantitative Biosciences Institute University of California San Francisco, 1700 4th Street, San Francisco, CA 94158)
478 2018 Proteins C

Marcin Tabaka and Aviv Regev (Broad Institute of MIT and Harvard, Cambridge, US)
479 2018 Transcripts C

Adin Aoki, Carlos Ayala, Lynn Agre, Rachana Tyagi, Wise Young (604 Allison Rd, Piscataway, NJ 08854)
480 2018 Cells C

Ayman Yousif, Nizar Drou, Jillian Rowe, Kristin Gunsalus (Center for Genomics and Systems Biology, NYU Abu Dhabi, Abu Dhabi UAE; NYU, New York, NY USA)
481 2018 Genomes C

Tony Di Sera, Chase Miller, Alistair Ward, Matt Velinder, Yi Qiao, Gabor Marth (University of Utah, Eccles Institute of Human Genetics, Salt Lake City, UT)
482 2018 Genomes D

Gustavo Salazar, Lorna Richardson, Neil Rawlings, Robert Finn (European Bioinformatics Institute)
483 2018 Genomes D

Christopher Fucile(1), Christopher Tipton(2), James Kobie(3), Inaki Sanz(2), Alexander Rosenberg(1) (University of Alabama at Birmingham, Birmingham AL, USA(1); Emory University, Atlanta GA, USA(2); University of Rochester Medical Center, Rochester NY, USA(3))
484 2018 Transcripts D

Andrea Winterbottom, Steve Trevanion, Imran Salam, Andy Yates, Paul Flicek (European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge, CB10 1SD, United Kingdom)
485 2018 Genomes D

Florian Gnad, Bin Zhang, Vidhisha Nandhikonda, Beth Murray, Jon M. Kornhauser, Vaughan Latham, Elzbieta Skrzypek, Peter V. Hornbeck (Cell Signaling Technology, Danvers, USA)
486 2018 Proteins D

György Papp, Roland Kunkli (University of Debrecen, Faculty of Informatics, Debrecen, Hungary)
487 2018 Genomes D

Xin-Yi Chua, James M. Hogan, Daniel Johnson (Queensland University of Technology, Brisbane, Australia)
488 2018 Populations D

Catherine Laplace (77 avenue Louis Pasteur, NRB 10, Boston, 02134)
489 2018 Art & Biology Y

Marwan Abdellah (Campus Biotech - Chemin des Mines 9, Geneva 1202, Switzerland)
490 2018 Art & Biology Y

Margot Riggi (Geneva University, Geneva, Switzerland)
491 2018 Art & Biology Y

Illustration: Valerie Altounian/Science; Data: RSG Group/Institut Pasteur; Data processing: J. Mozziconacci, R. Koszul, and J. D. Boeke (1200 NEW YORK AVE NW; Washington, DC 20005)
492 2018 Art & Biology Y

Kyle McClary, Trisha Williams (The Bridge Institute at USC, 1002 Childs Way, Los Angeles, CA 90089)
493 2018 Art & Biology Y

Maria Voigt (RCSB Protein Data Bank | Rutgers, The State University of New Jersey |Center for Integrative Proteomics Research | 174 Frelinghuysen Rd, Piscataway, NJ 08854-8076)
494 2018 Art & Biology Y

Lydia Gregg, MA, CMI, FAMI (Division of Interventional Neuroradiology, 1800 Orleans St., Bloomberg Bldg Rm 7218, Baltimore, MD 21287)
495 2018 Art & Biology Y

Adin M Aoki (604 Allison Rd, Piscataway, NJ 08854)
496 2018 Art & Biology Y

image: Matt Wimsatt, text: Kate Reed (The Jackson Laboratory for Genomic Medicine, 10 Discovery Drive, Farmington, CT 06032)
497 2018 Art & Biology Y

Bovaird, Samantha; Benoit Bouvrette, Louis Philip; Lécuyer, Eric (Institut de Recherches Clinique de Montréal, Montréal, Canada; Département de Biochimie, Université de Montréal, Montréal, Canada; Department of experimental medicine, McGill, Montréal, Canada)
498 2018 Art & Biology Y

Eric Weitz (Broad Institute of MIT and Harvard, Cambridge, MA, United States of America)
499 2018 Art & Biology Y


Sean O'Donoghue & James Procter (Garvan Institute, CSIRO Data61, and University of Dundee)
501 2017 Art & Biology Y

Sean O'Donoghue & James Procter (Garvan Institute, CSIRO Data61, and University of Dundee)
502 2017 Art & Biology Y


Florence Wang and Scott Chandry (CSIRO Australia)
504 2017 Genomes A

Kaspi A., Kobow K., Harikrishnan K. N., Ziemann M., Khurana I., Blümke I., & El-Osta A. (Department of Diabetes, Central Clinical School, Monash University, Melbourne, Australia; Department of Neuropathology, University Hospital Erlangen, Schwabachanlage, Germany)
505 2017 Genomes A

Maja Divjak, Adam Hunt, Ryan Granger, David Donovan, Jacinta Duncan (Gene Technology Access Centre, Melbourne, Australia; Dead on Sound, Melbourne, Australia; New Foundations Medical Mission, Cambridge, United Kingdom)
506 2017 Cells A

Aleona Swegen (University of Newcastle, Callaghan, NSW, Australia)
507 2017 Proteins A

Jieyi Zhao, Lingxi Zhou, Guocai Chen, Hua Xu, Jijun Tang, W. Jim Zheng (University of Texas Health Science Center at Houston)
508 2017 Genomes A

Merry Wang, Aaron Virshup, Eli Groban (Autodesk Life Sciences)
509 2017 Proteins A

Annelie Marquardt, Dana Pascovici, Frikkie Botha (Sugar Research Australia, Brisbane, Australia; Australian Proteome Analysis Facility, Sydney, Australia )
510 2017 Proteins B

Hieu T. Nim, Björn Sommer, Karsten Klein, Andrea Flack, Wolfgang Fiedler, Martin Wikelski and Falk Schreiber (Faculty of Information Technology, Monash University, Clayton, Australia; Department of Computer and Information Science, University of Konstanz, Konstanz, Germany)
511 2017 Populations B

John Salamon (1), Xiaoyan Qian (3), Mats Nilsson (3), David Lynn (1,2) ((1) South Australian Health and Medical Research Institute, Adelaide, Australia (2) Flinders University, Bedford Park, Australia (3) Stockholm University, Stockholm, Sweden)
512 2017 Genomes B

Justin Muir (Walter & Eliza Hall Institute, Melbourne, Australia)
513 2017 Other B

Rowland Mosbergen, Isha Nagpal, Othmar Korn, Ariane Mora, Tyrone Chen, Chris Pacheco Rivera and Christine A Wells (30 Royal Parade, Parkville VIC 3010, Australia)
514 2017 Transcripts B

Jim Procter, Mungo Carstairs, Kira Mourao, Charles Ofoegbu, Geoff Barton. (Barton Group, Division of Computational Biology, School of Life Sciences, University of Dundee, UK)
515 2017 Proteins B

Alvaro Berg Sotoa (1), Helene Marsh (1), Yvette Everingham (2), Joshua N. Smith (3), Guido J. Parra (4), Michael Noad (5) (James Cook University, Townsville, Australia; Murdoch University, Perth, Australia; Flinders University, Adelaide, Australia; University of Queensland, Brisbane, Australia)
516 2017 Populations B

Andrew Burgess, Jenny Vuong, Samuel Rogers, Marcos Malumbres, Seán I. O’Donoghue (Garvan Institute of Medical Research. 370 Victoria Street, Darlinghurst, NSW 2010)
517 2017 Cells B

Xun Zhu, Thomas Wolfgruber, Austin Tasato, Lana Garmire (University of Hawaii Cancer Center, 701 Ilalo St, Honolulu, HI 96813)
518 2017 Transcripts C

Shian Su, Charity Law, Matthew Ritchie (Walter and Eliza Hall Institute, 1G Royal Parade, Parkville 3052 Australia)
519 2017 Genomes C


Daniel Janies (University of North Carolina at Charlotte)
521 2017 Genomes C

Jenny Vuong, Benedetta Frida Baldi, Christopher Hammang, Seán I. O'Donoghue (384 Victoria Street)
522 2017 Other C

Benedetta Frida Baldi, Kenny Sabir, Sean O'Donoghue (The Garvan Institute of Medical Research, Darlinghurst, NSW; CSIRO-Data61,Eveleigh, NSW )
523 2017 Genomes C

Nicolette S. Birbara, James Otton, Nalini Pather (School of Medical Sciences, Medicine)
524 2017 Genomes C

Mirjam Figaschewski and Julian Heinrich (University of Tuebingen, Tuebingen, Germany)
525 2017 Proteins C

Ummul H Mohamad and Puteri NE Nohuddin (Institute of Visual Informatics, Universiti Kebangsaan Malaysia, 43600 UKM, Bangi, Selangor, Malaysia)
526 2017 Other A

Vladimir Naumov, Alexey Alexeev, Ivan Balashov, Vadim Lagutin, Pavel Borovikov (4, Oparin street, Moscow Russian Federation 117997)
527 2017 Genomes B

Annelie Marquardt, Dana Pascovici (Sugar Research Australia, Brisbane, Australia; Australian Proteome Analysis Facility, Sydney, Australia )
528 2017 Art & Biology Y

Anna Hupalowska (University of Cambridge, United Kingdom)
529 2017 Art & Biology Y

Benedetta Frida Baldi (The Garvan Institute of Medical Research, Darlinghurst, NSW)
530 2017 Art & Biology Y

Benjamin Leow (South Australian Health and Medical Research Institute, Adelaide, Australia; University of Adelaide, Adelaide, Australia)
531 2017 Art & Biology Y

Gabriel Aeschlimann (Ribosome Studio, Hünenberg, Switzerland; ZHAW, Wädenswil, Switzerland)
532 2017 Art & Biology Y

Ummul H Mohamad and Puteri NE Nohuddin (Institute of Visual Informatics (IVI), Universiti Kebangsaan Malaysia, 43600 UKM Bangi, Selangor, Malaysia)
533 2017 Art & Biology Y

Anna Liza Kretzschmar (Climate Change Cluster, University of Technology Sydney, Australia)
534 2017 Art & Biology Y

Maja Divjak, Adam Hunt, Ryan Granger, David Donovan, Jacinta Duncan (Gene Technology Access Centre, Melbourne, Australia; Dead on Sound, Melbourne, Australia; New Foundations Medical Mission, Cambridge, United Kingdom)
535 2017 Art & Biology Y

536 2017 Art & Biology Y


Garry W Lynch, Ana Paula Mello Lemgruber, Maria Rocha Costa, Stefanie Portelli, Christopher J Blasi, John S Sullivan, William Bret Church (Sydney University, School of Medicine & School of Veterinary Science. )
538 2017 Art & Biology Y

Leo Herson (University of Technology Sydney, Australia)
539 2017 Art & Biology Y
")
Sean O'Donoghue and James Procter (EMBL Heidelberg, Germany)
540 2016 Art & Biology Y
")
Chanaka Mannapperuma, David Sundell, Nicolas Delhomme, and Nathaniel Street (Umeå University)
541 2016 Genomes A

Adrien Villain, Christophe Bécavin and Pierre Lechat (Institut PASTEUR, PARIS, FRANCE)
542 2016 Genomes A

Stephen Taylor and Roger Noble (University of Oxford, Oxford, UK)
543 2016 Genomes A

Thomas Moerman, Dries Decap, Toni Verbeiren, Jan Fostier, Joke Reumers, Jan Aerts (VDA-lab, ESAT/STADIUS, KU Leuven, Belgium – iMinds; INTEC, UGent, Belgium – iMinds; Janssen R&D, Beerse, Belgium; ExaScience Life Lab, Leuven, Belgium)
544 2016 Genomes A

Julian Heinrich, Jenny Vuong, Sandeep Kaur, Sean I. O'Donoghue (CSIRO, Sydney, Australia; UNSW, Sydney, Australia; The Garvan Institute of Medical Research, Sydney, Australia)
545 2016 Proteins A

Khan, M. J, Figueiredo, L.T., Aquino.V.H (University of Sao Paulo, Ribeirao Preto , Sao Paulo, Brazil)
546 2016 Genomes A

Monika Balvočiūtė and Daniel H. Huson (University of Tübingen, Faculty of Computer Science, Algorithms in Bioinformatics, Room C310b, Sand 14, 72076 Tübingen, GERMANY )
547 2016 Other A

Georg Tremmel, Paul Sheridan, Atsushi Niida (Laboratory for DNA Information Analysis, Institute of Medical Science, University Of Tokyo, 4-6-1 Shirokanedai, Tokyo 108-8639, Japan)
548 2016 Other A

James Dunbar, Konrad Krawczyk, Jinwoo Leem, Claire E. Marks, Jaroslaw Nowak, Cristian Regep, Guy Georges, Sebastian Kelm, Bojana Popovic, Charlotte M. Deane (University of Oxford, Oxford, UK)
549 2016 Proteins A

Colin Combe, Marine Dumouseau, Josh Heimbach, Birgit Meldal, Sandra Orchard, Juri Rappsilber (Wellcome Trust Centre for Cell Biology, School of Biological Sciences, University of Edinburgh, Edinburgh EH9 3BF, United Kingdom)
550 2016 Proteins A

Davide Heller, Alexander Tournier, Lorenzo Gatti, Andreas Hoppe, Simon Restrepo, Nicolas Tapon, Konrad Basler, Yanlan Mao (UZH, Zurich, Switzerland; ZHAW, Zurich, Switzerland; SIB, Lausanne, Switzerland; UCL, London, United Kingdom; Kingston University, Kingston, United Kingdom; Cancer Research UK, London, United Kingdom;)
551 2016 Organisms A

Christina Rode, Marella de Bruijn (Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, UK)
552 2016 Organisms A

Christian Stolte, Sergei German, Kevin Shi, Sudeep Mehrotra, Dimitrije Jevremovic, Nina Lapchyk, Avinash Abhyankar, Ann-Katrin Emde, Nathaniel Pearson, Karen Hackett, Shailu Gargeya, and Toby Bloom (New York Genome Center, New York, USA)
553 2016 Genomes A

Loic Royer, Martin Weigert, Ulrik Günther, Nicola Maghelli, Florian Jug, Ivo F. Sbalzarini, Eugene W. Myers (Center for Systems Biology & Max Planck Institute for Molecular Cell Biology and Genetics, Dresden, Germany; Technische Universität Dresden, Dresden, Germany)
554 2016 Other A


Andreas Göransson, Gustav Bohlin and Lena Tibell (Dep. of Science and Technology, Linköping University / C-Research Visualization Center C, Norrköping, Sweden)
556 2016 Genomes A

Alyssa Morrow, Eric Tu, Frank Austin Nothaft, Anthony D. Joseph, David A. Patterson (387 Soda Hall, Berkeley, CA 94720 United States)
557 2016 Genomes B

David Ma, Christian Stolte, James Krycer, David James, Seán O'Donoghue (Garvan Institute of Medical Research, Sydney, Australia; CSIRO, Sydney, Australia.)
558 2016 Cells B

Ilinca Tudose, Nathalie Conte, Jeremy Mason, Terry Meehan (European Bioinformatics Institute, EMBL-EBI, Hinxton, UK)
559 2016 Other B

Sara El-Gebali 1, Nancy George 1, Robin Liechti 2, Lou Götz 2, Isaac Crespo 2, Ioannis Xenarios 2, Thomas Lemberger 1 (1-EMBO, Heidelberg, Germany, 2-Vital-IT, SIB Swiss Institute of Bioinformatics, Switzerland)
560 2016 Other B

Stephen Todd, Peter Todd, Frederic Fol Leymarie, William Latham, Jim Hughes, Stephen Taylor (Goldsmiths, University of London, U.K.; University of Oxford, U.K.)
561 2016 Proteins B

Ibrahim Tanyalcin, Carla Al Assaf, Alexander Gheldof, Katrien Stouffs, Willy Lissens, Anna C. Jansen (VUB, Brussels, Belgium; KUL, Leuven, Belgium; UZ Brussel, Brussels, Belgium; UZ Brussel, Brussels, Belgium; UZ Brussel, Brussels, Belgium; UZ Brussel, Brussels, Belgium)
562 2016 Proteins B

David Bouvier, Luis Salamanca, Rudi Balling and Alexander Skupin. (Luxembourg Centre of Systems Biomedicine, University of Luxembourg, Belval, Luxembourg)
563 2016 Cells B

Juha Karjalainen (University Medical Center Groningen, Groningen, The Netherlands)
564 2016 Genomes B

Michael Zyracki, Joseph Schaeffer, Andrew Kimoto, Malte Tinnus, Peter Jones, Dave Parker, Merry Wang (Bio/Nano, Autodesk Research, Toronto, Canada)
565 2016 Proteins B

Jasmin Imran Alsous, Paul Villoutreix, and Stanislav Shvartsman (Princeton University, NJ, USA)
566 2016 Organisms B

Fidel Ramírez, Vivek Bhardwaj, Thomas Manke (Max Planck Institute of Immunobiology and Epigenetics, Stübeweg 51, D-79108 Freiburg, Germany)
567 2016 Genomes B

Benedikt Rauscher, Florian Heigwer, Michael Boutros (Deutsches Krebsforschungszentrum, Division Signaling and Functional Genomics, Im Neuenheimer Feld 580, Heidelberg, Germany)
568 2016 Genomes B

Arnim Jenett, Pierre Affaticati, Elodie de Job, Laurie Riviere, Barbara Rizzi, Johanna Djian-Zaouche, Jean-Stephane Joly (Tefor Core Facility, Paris-Saclay Institute of Neuroscience, CNRS, Université Paris-Saclay, 91190 Gif-sur-Yvette, France)
569 2016 Genomes B

Thomas SB Schmidt, João F Matias Rodrigues, Christian von Mering (Institute of Molecular Life Sciences, University of Zurich)
570 2016 Genomes C

Beat Wolf 1,2, Pierre Kuonen 1, Thomas Dandekar 2 (1 University of Applied Sciences Western Switzerland, Fribourg, Switzerland;2 University of Würzburg, Germany)
571 2016 Genomes C

Maria Voigt, Shuchismita Dutta, David S. Goodsell, Christine Zardecki, Stephen K. Burley and the entire RCSB PDB Team (Rutgers, The State University of New Jersey Center for Integrative Proteomics Research 174 Frelinghuysen Rd Piscataway, NJ 08854-8076 )
572 2016 Proteins C

N. Perdigão, J. Heinrich, C. Stolte, K. S. Sabir, M. J. Buckley, B. Tabor, B. Signal, B. S. Gloss, C. J. Hammang, B. Rost, A. Schafferhans, S. I. O'Donoghue (Instituto Superior Técnico - Universidade de Lisboa; Commonwealth Scientific and Industrial Research Organisation (CSIRO); Garvan Institute of Medical Research; Technische Universität München)
573 2016 Genomes C

Jun Seita, Irving L. Weissman (Institute for Stem Cell Biology and Regenerative Medicine, Stanford University, Stanford, CA, 94305, USA)
574 2016 Transcripts C

Kamil Erguler and the OrthoMap Ninjas (The Cyprus Institute, Nicosia, Cyprus)
575 2016 Genomes C

Michael T. Hons, Pim J. Huis in ‘t Veld, Jan Kaesler, Franz Herzog, Alexander Schleiffer, Jan-Michael Peters & Holger Stark (MPI for Biophysical Chemistry, Am Faßberg 11, 37077 Göttingen)
576 2016 Genomes C

Niklas Biere, Anja Doebbe, Daniel Jäger, Nils Rothe, Sebastian Goldenberg, Benjamin Friedrich, Christoph Brinkrolf, Benjamin Kormeier, Olaf Kruse, Björn Sommer (Bielefeld University/MPI for the Physics of Complex Systems, Dresden/Monash University, Melbourne)
577 2016 Genomes C

Nikiforos Karamanis, Francis Rowland, Miguel Pignatelli and the CTTV Consortium (Center for Therapeutic Target Validation (CTTV), Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, UK)
578 2016 Other C

James Hadfield, Simon Harris (Wellcome Trust Sanger Institute, Cambridge, UK)
579 2016 Genomes C

Daniel Alcaide, Masoud Zamani Esteki, Amin Ardeshirdavani, Yves Moreau, Thierry Voet, Jan Aerts (KU Leuven, Leuven, Belgium)
580 2016 Genomes C

Nicola Lopizzo, Veronica Begni, Sarah Tosato, Simona Tomassi, Marco Andrea Riva, Carmine Maria Pariante, Annamaria Cattaneo (IRCCS Fatebenefratelli San Giovanni di Dio, Brescia; Response variability to Psychotropic, Paris, France; University of Milan, Milan, Italy; University of Verona, Verona, Italy; King’s College, London)
581 2016 Transcripts C

Stuart G. Jantzen, Jodie Jenkinson, Gaël McGill (Biomedical Communications Unit, Department of Biology, University of Toronto, Mississauga, Ontario; Center for Molecular and Cellular Dynamics, Harvard Medical School, Boston, Massachusetts)
582 2016 Proteins C

Paul Brennan (School of Medicine, Cardiff University, Heath Park, Cardiff, CF14 4SZ, United Kingdom, )
583 2016 Proteins C

Iliona Wolfowicz, Annika Guse (Centre for Organismal Studies, Im Neuenheimer Feld 329, 69120 Heidelberg, Germany)
584 2016 Organisms C

Bill Hill and Richard A. Baldock (MRC Human Genetics Unit, IGMM, University of Edinburgh)
585 2016 Organisms D

Ed Griffiths & Michael Gray (Wellcome Trust Sanger Institute, Hinxton, Cambridgeshire, UK)
586 2016 Genomes D

Chris Hammang, Sean O'Donoghue, Peter Dodds (384 Victoria Street, Darlinghurst NSW 2010)
587 2016 Genomes D

Stephen Todd, Frederic Fol Leymarie, William Latham, Peter Todd, Pedro Quijada Leyton, Andy Thomason (Goldsmiths, University of London)
588 2016 Genomes D

Graham Johnson & Greg Johnson (Allen Institute for Cell Science, Seattle, USA)
589 2016 Cells D

Benedetta Frida Baldi (EMBL-EBI, Cambridge, UK; University of Cambridge, Cambridge,UK)
590 2016 Other D

Ioannis Kirmitzoglou, Robert M. MacCallum & George K. Christophides (Imperial College London)
591 2016 Populations D

Satoshi Nishimura ( Center for Molecular Medicine, Jichi Medical University, Tochigi, Japan, and 2 Department of Cardiovascular Medicine, the University of Tokyo, Tokyo, Japan)
592 2016 Cells D

Jansi Thiyagarajan, Jan Aerts (KU Leuven, Leuven, Belgium)
593 2016 Other D

Volker Hilsenstein, Sabine Reither, Beate Neumann, Christian Tischer, Rainer Pepperkok (EMBL Advanced Light Microscopy Facility, Heidelberg, Germany)
594 2016 Genomes D
")
Andrii Iudin, Paul Korir, Ingvar Lagerstedt, Jose Salavert-Torres, Eduardo Sanz-García, Vladislav Lysenkov, Ardan Patwardhan, Gerard Kleywegt (EMBL-EBI, Wellcome Genome Campus Hinxton, Cambridge CB10 1SD, United Kingdom)
595 2016 Proteins D

Andy Thomason, Pedro Quijada Leyton, Ioannis Filippis, Michael Sternberg, William Latham, Frederic Fol Leymarie (Goldsmiths College, New Cross, London.)
596 2016 Proteins D

Anil S Thanki, Nicola Soranzo, Robert P. Davey (The Genome Analysis Centre, Norwich Research Park, Norwich, UK)
597 2016 Genomes D

Abhishek Kumar, Narendar Kolimi#, L Ponoop P Patro# and Thenmalarchelvi Ratinavelan* (Department of Biotechnology, Indian Institute of Technology Hyderabad, Kandi, Telangana, India-502285.)
598 2016 Other A

Bharathi Reddy Kunduru†, Sanjana Anilkumar Nair†, Shivangi Sachdeva# and Thenmalarchelvi Rathinavelan* (Department of Biotechnology, Indian Institute of Technology Hyderabad, Kandi, Telangana, India-502285.)
599 2016 Genomes B

JM Hogan, M. Brereton, D Johnson, M. Rittenbruch, S O'Donoghue, C Hammang, J Vuong, J. Heinrich, D Syme (Queensland University of Technology, CSIRO/Garvan Institute, MSR)
600 2016 Genomes B

Christos Christoforos, Luisa Lorenzi MD, Fabio Facchetti MD, PhD (Pathology Unit Department of Molecular and Translational Medicine University of Brescia Spedali Civili di Brescia 25123 Brescia Italy )
601 2016 Cells C

Andrea Schafferhans, Kenneth Sabir, Maximilian Hecht, Burkhard Rost, Sean O'Donoghue (Technische Universität München, Germany; Garvan Institute of Medical Research, Sydney, Australia; CSIRO Computational Informatics, Sydney, Australia)
602 2016 Proteins D

Beat Wolf 1,2, Pierre Kuonen 1, Thomas Dandekar 2 (University of Applied Sciences Western Switzerland, Fribourg, Switzerland; University of Würzburg, Germany)
603 2016 Art & Biology Y

Abhishek Kumar, Narendar Kolimi#, L Ponoop P Patro# and Thenmalarchelvi Ratinavelan* (Department of Biotechnology, Indian Institute of Technology Hyderabad, Kandi, Telangana, India-502285.)
604 2016 Art & Biology Y

Bharathi Reddy Kunduru†, Sanjana Anilkumar Nair†, Shivangi Sachdeva# and Thenmalarchelvi Rathinavelan* (Department of Biotechnology, Indian Institute of Technology Hyderabad, Kandi, Telangana, India-502285.)
605 2016 Art & Biology Y

Andreas Göransson (Dep. of Science and Technology, Linköping University / C-Research Visualization Center C, Norrköping, Sweden)
606 2016 Art & Biology Y

Jasmin Imran Alsous (Princeton University, NJ, USA)
607 2016 Art & Biology Y

Maria Voigt, David S. Goodsell (RCSB Protein Data Bank, Rutgers, The State University of New Jersey Center for Integrative Proteomics Research 174 Frelinghuysen Rd Piscataway, NJ 08854-8076)
608 2016 Art & Biology Y

Stephen Todd, Frederic Fol Leymarie, William Latham, Peter Todd, Pedro Quijada Leyton, Andy Thomason (Goldsmiths, University of London)
609 2016 Art & Biology Y

Ibrahim Tanyalcin, Carla AL Assaf (VUB, Brussels, Belgium; KUL, Leuven, Belgium)
610 2016 Art & Biology Y

Barth van Rossum, Leif Schroeder (Leibniz-Institut fuer Molekulare Pharmakologie (FMP), Berlin, Germany)
611 2016 Art & Biology Y

David Bouvier and Luis Salamanca. (Luxembourg Centre of Systems Biomedicine, University of Luxembourg, Belval, Luxembourg)
612 2016 Art & Biology Y

Georg Tremmel (Laboratory for DNA Information Analysis, Institute of Medical Science, University Of Tokyo, 4-6-1 Shirokanedai, Tokyo 108-8639, Japan)
613 2016 Art & Biology Y

Stuart Jantzen (Biomedical Communications Unit, Department of Biology, University of Toronto, Mississauga, Ontario)
614 2016 Art & Biology Y

Peter Todd, Stephen Todd, Frederic Fol Leymarie, William Latham (Goldsmiths, University of London, U.K.)
615 2016 Art & Biology Y

Peter Todd, Stephen Todd, Frederic Fol Leymarie, William Latham, Stephen Taylor, Jim Hughes (Goldsmiths, University of London)
616 2016 Art & Biology Y

Christina Rode (Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, UK)
617 2016 Art & Biology Y

Fidel Ramírez (Max Planck Institute of Immunobiology and Epigenetics, Stübeweg 51, D-79108 Freiburg, Germany)
618 2016 Art & Biology Y

William Latham, Frederic Fol Leymarie, Andy Thomason, Pedro Quijada Leyton, Michael Sternberg, Ioannis Filippis (Goldsmiths, University of London, and Imperial College London)
619 2016 Art & Biology Y

Iliona Wolfowicz (Centre for Organismal Studies, Im Neuenheimer Feld 329, 69120 Heidelberg, Germany)
620 2016 Art & Biology Y

Wen-ti Liu (Max Planck Institute for Biophysical Chemistry Am Faßberg 11, 37077 Göttingen, Germany)
621 2016 Art & Biology Y


Christine Molenda (Institute for Cell Biology and Neuroscience, Department of Developmental Biology of Vertebrates, Goethe-Universität - Campus Riedberg, Max-von-Laue-Straße 13. D-60438 Frankfurt am Main)
623 2016 Art & Biology Y

Christian Stolte (New York Genome Center, New York, USA)
624 2016 Art & Biology Y

Haruko Hirukawa, Kimi Ogasawara (Institute of Transformative Bio-Molecules (ITbM), Nagoya University Furo-cho, Chikusa-ku, Nagoya 464-8601, Japan)
625 2016 Art & Biology Y

Arnim Jenett, Pierre Affaticati, Elodie de Job, Laurie Riviere, Barbara Rizzi, Johanna Djian-Zaouche, Jean-Stephane Joly (Tefor Core Facility, Paris-Saclay Institute of Neuroscience, CNRS, Université Paris-Saclay, 91190 Gif-sur-Yvette, France)
626 2016 Art & Biology Y

Sean O'Donoghue, James Procter, Bang Wong, Christian Stolte (Broad Institute of MIT and Harvard)
627 2015 Art & Biology Y

Boris Fedorov, Leonid Zaslavsky and Tatiana Tatusova (National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA)
628 2015 Genomes A

Dan Tulpan, Serge Léger (Information and Communication Technologies, National Research Council, Canada)
629 2015 Genomes A

Eric van der Helm, Henrik Marcus Geertz-Hansen Hans Jasper Genee, Sailesh Malla, and Morten O. A. Sommer (Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Hørsholm, Denmark)
630 2015 Genomes A

Arnaud Ceol and Heiko Muller (Center for Genomic Science of IIT@SEMM, Fondazione Istituto Italiano di Tecnologia (IIT), Via Adamello 16, 20139 Milan, Italy)
631 2015 Genomes A

Tauno Metsalu, Jaak Vilo (University of Tartu, Tartu, Estonia)
632 2015 Transcripts A

Derek P. Ng, Michael Corrin, Jodie Jenkinson, Graham Johnson (Biomedical Communications, Dept. Biology, University of Toronto, Toronto, Canada; California Institute for Quantitative Biosciences, University of California, San Francisco, USA )
633 2015 Proteins A

Stuart G. Jantzen, Jodie Jenkinson, Gaël McGill (Biomedical Communications Unit, Department of Biology, University of Toronto, Mississauga, Ontario; Center for Molecular and Cellular Dynamics, Harvard Medical School, Boston, Massachusetts )
634 2015 Other A

Alberto I. Roca, Isabella Villano, Aaron C. Abajian (ProfileGrid.org, Irvine, CA, USA)
635 2015 Genomes A

Christian Stolte, Kenny Sabir, Sean O'Donoghue, Maria Kalemanov, Benjamin Wellmann, Vivian Ho, Fabian Buske, Manfred Roos, Nelson Perdigao, Julian Heinrich, Burkhard Rost, Andrea Schafferhans (CSIRO Digital Productivity, Sydney, Australia)
636 2015 Proteins A

Jia-Ming Chang, Giacomo Cavalli (Institute of Human Genetics (IGH), UPR 1142, CNRS 141, rue de la Cardonille, 34396 Montpellier Cedex 5, France)
637 2015 Genomes A

Kalle Leinonen, Suzanne Paquette, and William Longabaugh (Institute for Systems Biology, Seattle, WA USA)
638 2015 Genomes B

Raivo Kolde, Jaak Vilo (University of Tartu, Tartu, Estonia; Broad Institute, Cambridge, USA)
639 2015 Transcripts B

Michael Gray (Wellcome Trust Sanger Institute, Cambridge, England)
640 2015 Genomes B

Ryo Sakai, Raf Winand, Toni Verbeiren, Andrew Vande Moere, Jan Aerts (Department of Electrical Engineering (ESAT) STADIUS Center for Dynamical Systems, Signal Processing and Data Analytics, KU Leuven, 3001, Belgium. iMinds Medical IT, KU Leuven, 3001, Belgium)
641 2015 Genomes B

Catherine Laplace (Benoist-Mathis Lab, Dept. of Microbiology & Immunobiology 77, Avenue Louis Pasteur . HMS NRB - Room1052K Boston, MA 02115 United States)
642 2015 Transcripts B

Nicolas F. Fernandez, Matthew R. Jones, Greg W. Gundersen, Qiaonan Duan, Andrew D. Rouillard, Joel T. Dudley, and Avi Ma’ayan (Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, New York, NY 10029 USA )
643 2015 Cells B

Diego M S, Ning J, Aaron B, Ricardo Z. N. V, Nitin S B (Institute for Systems Biology, Baliga's Lab, Seattle, USA; LabPIB, Department of Computing and Mathematics FFCLRP-USP, University of Sao Paulo, Ribeirao Preto, Brazil;)
644 2015 Genomes B

Chris Hammang, Drew Berry, Christian Stolte, Olivier Salvado, William Wilson, Sean O'Donoghue (384 Victoria St, Darlinghurst, NSW, Australia, 2010)
645 2015 Proteins B

Xavier Watkins (EMBL-European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridgeshire CB10 1SD, United Kingdom)
646 2015 Transcripts B

Angela Makolo, Raphael D. Isokpehi (of Computer Science, University of Ibadan, Nigeria;College of Science, Engineering and Mathematics, Bethune-Cookman University, Daytona Beach, FL, USA)
647 2015 Genomes B

Grace Hsu, Evelyn Maizels, Deb Milkowski (University of Illinois at Chicago, Chicago, IL, USA)
648 2015 Proteins C

Samuel Hertig, Graham T. Johnson, Thomas D. Goddard, Thomas E. Ferrin (University of California, San Francisco)
649 2015 Proteins C

Patricio Yankilevich, Maximiliano de Sousa Serro, Daniel Koile (Instituto de Investigación en Biomedicina de Buenos Aires (IBioBA) - CONICET - Partner Institute of the Max Planck Society, Buenos Aires, Argentina)
650 2015 Genomes C

Liying Zhang, Veer Shanmugasundaram, and Robert Stanton (Pfizer Inc)
651 2015 Other C

Sandra R. Schachat (Department of Paleobiology, Smithsonian Institution)
652 2015 Genomes C

Zhi Li, Merry Wang, Shannon Roberts, Valera Castanov, Jacobo Bibliowicz, Jeremy Mogk, Azam Khan, Anne Agur (University of Toronto, Medical Sciences Building, Rm. 1158, 1 King's College Circle, Toronto, ON, M5S 1A8, Canada)
653 2015 Organisms C

Curtis Lisle, Jeffrey Baumes, Anthony Wehrer, Luke Harmon, Robert W. Thacker, Chelsea Specht, Charles Hughes, Jorge Soberon (KnowledgeVis, LLC, Maitland, FL, USA)
654 2015 Populations C

Bálint Antal, Anatole Chessel, and Rafael E. Carazo Salas ( Genetics Department, University of Cambridge, Downing Street, Cambridge, CB2 3EH, United Kingdom )
655 2015 Populations C

Kenneth Sabir, Dr Fabian Buske, Christian Stolte, Prof. Seokhee Hong, Prof. Susan Clark, Dr Sean O’Donoghue (Garvan Institute of Medical Resaerch, Sydney, Australia, University of Sydney, Sydney, Australia)
656 2015 Genomes C

Jennifer Frazier, Joyce Ma, Kwan-Liu Ma (The Exploratorium, San Francisco, CA; The UC Davis Center for Visualization, Davis, CA)
657 2015 Other C

Andreas Maunz, Moritz Gilsdorf, Sebastien Fournier, Christian Blumenröhr, Ralf Horstmöller, Jörg Schmiedle (Hoffmann-La Roche AG Innovation Center Basel, Switzerland)
658 2015 Transcripts D

Roy Williams and Amar Das (Dartmouth College, Hanover, New Hampshire)
659 2015 Genomes D

Martijn P. van Iersel, Joanna L. Parmley (General Bioinformatics Limited, Reading, UK)
660 2015 Cells D

Qiaonan Duan, Avi Ma'ayan (Icahn School of Medicine at Mount Sinai, New York, USA)
661 2015 Genomes D

Keiichiro Ono (UC, San Diego )
662 2015 Cells D

Anurag Verma, Scott Dudek, Marylyn Ritchie, Sarah Pendergrass (512 Wartik Lab, Pennsylvania State University)
663 2015 Genomes D

Helen Cook (University of Copenhagen, Copenhagen, Denmark)
664 2015 Proteins D

David K.G. Ma, Christian Stolte, Sandeep Kaur, Michael Bain, and Seán I. O’Donoghue (384 Victoria St Darlinghurst NSW 2010 Australia)
665 2015 Genomes D

Neslisah Torosdagli, Sumanta Pattanaik, Curtis Lisle (Institute 1: Dept. of EECS (CS Division) University Of Central Florida,Orlando, FL, USA; Institute 2: KnowledgeVis LLC, Maitland, FL, USA.)
666 2015 Other A

Grace Hsu, Evelyn Maizels, Deb Milkowski (University of Illinois at Chicago, Chicago, IL, USA)
667 2015 Art & Biology Y

Catherine Laplace (Benoist-Mathis Lab, Dept. of Microbiology & Immunobiology 77, Avenue Louis Pasteur . HMS NRB - Room1052K Boston, MA 02115 United States)
668 2015 Art & Biology Y

Kristin Johnson and Jason Dearling, PhD (Vascular Biology Program, Boston Children's Hospital, 1 Blackfan Street, Karp 12.112, Boston, MA, USA 02130)
669 2015 Art & Biology Y

Stuart Jantzen (Biomedical Communications Unit, Department of Biology, University of Toronto, Mississauga, Ontario)
670 2015 Art & Biology Y

Helen Cook (University of Copenhagen, Copenhagen, Denmark)
671 2015 Art & Biology Y

Xavier Watkins (EMBL-European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridgeshire CB10 1SD, United Kingdom)
672 2015 Art & Biology Y

Stefanie Kuzmiski (3-3040 Dundas Street West, Toronto, Ontario, Canada, M4E 2N3)
673 2015 Art & Biology Y

Evan Ingersoll & Gael McGill (Digizyme Inc, Brookline MA)
674 2015 Art & Biology Y

Luke Wilson, Jim Procter, Suzanne Duce and Geoff Barton. (College of Life Sciences and Duncan of Jordanstone College of Art and Design, University of Dundee, Dundee, Scotland, UK)
675 2015 Art & Biology Y

Patricio Yankilevich (Instituto de Investigación en Biomedicina de Buenos Aires (IBioBA) - CONICET - Partner Institute of the Max Planck Society, Buenos Aires, Argentina)
676 2015 Art & Biology Y

Sean O'Donoghue, James Procter, Christian Stolte (EMBL Heidelberg, Germany)
677 2014 Art & Biology Y

Bang Wong (Broad Institute of MIT and Harvard)
678 2014 Art & Biology Y

Nuchnoi P, Ohashi J, Naka I, Hananantachai H, Tokunaga K, Patarapotikul J (1 Faculty of Medical Technology, Mahidol University, Bangkok, Thailand)
679 2014 Genomes A

Ryo Sakai, Peter Konings, Yves Moreau, Koen Vanmechelen, Jan Aerts (KU Leuven, Leuven, Belgium; iMinds Medical IT, Leuven, Belgium)
680 2014 Genomes A

Nick R. Love (RIKEN:Center for Developmental Biology)
681 2014 Cells A

David E. Breen, Thomas Widmann, Linge Bai, Liyuan Sui, Frank Jülicher, Christian Dahmann (Drexel University, Philadelphia, PA, USA; GENYO, Granada, Spain; MPI-PKS, Dresden, Germany; TU Dresden, Germany)
682 2014 Cells A

Beat Wolf(1), Pierre Kuonen(1), Thomas Dandekar(2) ((1)University of Applied Sciences Western Switzerland, Fribourg, Switzerland; (2)University of Würzburg, Würzburg, Germany)
683 2014 Genomes A

Holly M. Bik (University of California, Davis, One Shields Ave, Davis, CA 95616, United States of America)
684 2014 Populations A

Pauline Gloaguen, Gilles Curien, Sylvain Bournais, Christophe Bruley, Florence Combes, Marianne Tardif, Yves Vandenbrouck, Giovanni Finazzi, Myriam Ferro and Norbert Rolland (CNRS, UMR 5168, CEA, DSV, iRTSV, PCV, 17 rue des Martyrs, F-38054 Grenoble, France-INSERM, EDyP, BGE/U1038, INSERM/CEA/Université Grenoble Alpes, F-38054 Grenoble France)
685 2014 Cells A

Dario Vianello, Federica Sevini, Claudio Franceschi (DIMES, Via San Giacomo n°12, Bologna, Italy)
686 2014 Genomes A

Gordon Stephen, Iain Milne, Micha Bayer, Paul D. Shaw, Sebastian Raubach, Sarah Hearne, Sukhwinder Singh, Peter Wenzl, David Marshall (The James Hutton Institute, Dundee, Scotland; International Maize and Wheat Improvement Center (CIMMYT), Texcoco, Mexico)
687 2014 Populations A

Maria Vila-Casadesús, Jan Graffelman, Meritxell Gironella, Juan José Lozano (Bioinformatics Platform CIBERehd, Barcelona, Spain; Gastrointestinal & Pancreatic Oncology Team CIBERehd/IDIBAPS/H.C.B., Barcelona, Spain; Department of Statistics and O.R. UPC, Barcelona, Spain)
688 2014 Transcripts A

Florian Heigwer, Chrisian Volz, Michael Boutros (German Cancer Research Center, Heiderlberg, Germany; Heidelberg University, Heidelberg, Germany)
689 2014 Genomes A

Abbeloos, R., Dhondt, S., Wuyts, N. and Inzé, D. (VIB Department of Plant Systems Biology, Technologiepark 927, 9052 Gent, BELGIUM)
690 2014 Organisms A

Seán I. O’Donoghue, Kenneth Sabir, Maria Kalemanov, Christian Stolte, Benjamin Wellmann, Vivian Ho, Fabian Buske, Manfred Roos, Nelson Perdigão, Julian Heinrich, Burkhard Rost, Andrea Schafferhans (CSIRO Computational Informatics, Sydney, Australia)
691 2014 Proteins A

Kenneth Sabir, Seán I. O’Donoghue, Maria Kalemanov, Christian Stolte, Benjamin Wellmann, Vivian Ho, Fabian Buske, Manfred Roos, Nelson Perdigão, Julian Heinrich, Burkhard Rost, Andrea Schafferhans (Garvan Institute of Medical Research, Sydney, Australia)
692 2014 Proteins A

Janos Binder, Sune Pletscher-Frankild, Kalliopi Tsafou, Christian Stolte, Seán I. O’Donoghue, Reinhard Schneider, Lars Juhl Jensen (European Molecular Biology Laboratory (EMBL), Heidelberg, Germany; Bioinformatics Core Facility, Luxembourg Centre for Systems Biomedicine (LCSB), Esch-sur-Alzette, Luxembourg)
693 2014 Genomes A

Christopher Hammang, Sean O'Donoghue, Christian Stolte, Drew Berry, Kenny Sabir, Kate Patterson, David Topping, Trevor Lockett, Julie Clarke (CSIRO, 11 Julius Avenue, North Ryde, Sydney, NSW, 2113)
694 2014 Genomes A

Hiroshi Ro , Kyoko Shinya, Kazuto Tominaga, Yasuhiro Suzuki (Nagoya Univ., Information and Science, Nagoya, Japan; Kobe Univ., Grad. school of Med, Kobe, Japan, Kano InfoNet, Matsumoto, Japan; Keio Univ., Grad. School of SDM, Yokohama, Japan)
695 2014 Genomes A

Dmitry Shurov, Grigory Armeev, Alexey Shaytan, Konstantin Shaitan (Lomonosov Moscow State University, Moscow, Russian Federation)
696 2014 Genomes B

Rowland Mosbergen,Othmar Korn,Jarny Choi,Nick Seidenman,Nick Matigian,Christine Wells (Australian Institute of Bioengineering and Nanotechnology,Brisbane,Australia;Walter & Eliza Hall Institute of Medical Research, Melbourne, Victoria, Australia)
697 2014 Genomes B

Rafal D. Urniaz (Medical University of Lublin ,Lublin, Poland)
698 2014 Proteins B

Catherine Leroy (Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge. CB10 1SA UK)
699 2014 Genomes B

Audrey M. Michel, Gearoid Fox, Anmol Kiran, Christof De Bo, Patrick B.F. O’Connor, Stephen M. Heaphy, James P.A. Mullan, Claire A. Donohue, Romika Kumari, Desmond G. Higgins, Pavel V. Baranov (Biochemistry Department, University College Cork, Ireland; Conway Institute, University College Dublin, Ireland; HOWEST University College West Flanders, Bruges, Belgium)
700 2014 Genomes B

Stefan Henrich, Jonathan Fuller, Michael Martinez, Stefan Richter, Rebecca Wade (HITS gGmbH, Schloss-Wolfsbrunnenweg 35, D-69118 Heidelberg)
701 2014 Proteins B

Stephen Taylor, Roger Noble, Samuel Conway (Weatherall Institute of Molecular Medicine, Oxford, United Kingdom; Coritsu Labs, Adelaide, Australia)
702 2014 Other B

Fidel Ramírez, Friederike Dündar, Sarah Diehl, Björn A. Grüning, Thomas Manke (Max-Planck-Institute for Immunobiology and Epigenetics, Freiburg, Germany, Department of Computer Science, University of Freiburg, Germany)
703 2014 Genomes B

Mireia Vilardell, Andrew worth, Maurice Whelan (European Comission, DG Joint Research Center, Intitute of Health and Consumer Protection, via E. Fermi 2749 Ispra, Italy)
704 2014 Genomes B

Joana Silva, Isabel F. Amaral, Pedro Quelhas (INEB - Instituto de Engenharia Biomédica, Universidade do Porto, Portugal)
705 2014 Other B

Ryo Sakai, Justin Ashworth, Jan Aerts (KU Leuven, Leuven, Belgium; iMinds Medical IT, Leuven, Belgium; Institute for Systems Biology, Seattle, USA;)
706 2014 Proteins B

David Rio Deiros, M Raveendran, G Fawcett, RA Gibbs and J Rogers (One Baylor Plaza, Houston, Texas 77030)
707 2014 Genomes B

Heng-Chang Chen, Arbert Jordan, Guillaume Filion (Centre for Genomic Regulation (CRG), Edif. PRBB, Dr. Aiguader, 88, 08003, Barcelona, Spain )
708 2014 Genomes B

Paula González Avalos, Lucas Schütz, Silvia Urbansky, Stefan Günther, Heike Leitte, Steffen Lemke (Im Neuenheimer Feld 230)
709 2014 Organisms B

Heba Sailem, Miguel Sanchez-Alvarez, Julia Sero, Chris Bakal (London, UK)
710 2014 Cells B

Eric van der Helm, Morten Sommer (The Novo Nordisk Foundation Center for Biosustainability (CFB) at the Technical University of Denmark, Hørsholm, Denmark)
711 2014 Genomes B

Anil S. Thanki, Xindong Bian, Robert P. Davey (The Genome Analysis Centre, Norwich Research Park, Norwich, UK)
712 2014 Genomes B

Martina Kutmon, Anwesha Bohler, Susan L. Coort, Alexander R. Pico, Chris T. Evelo (Department of Bioinformatics-BiGCaT, Maastricht University, The Netherlands; Gladstone Institute of Cardiovascular Disease, San Francisco, California, USA)
713 2014 Other B

Jelena Chuklina, Maxim Imakaev, Elena Evguenieva-Hackenberg, Mikhail Gelfand (Moscow Institute of Physics and Technology, Moscow, Russa; Institute for Information Transmission Problems, Moscow, Russia; MIT, Boston, USA)
714 2014 Genomes C

Jim Procter, Suzanne Duce, Geoff Barton and the Jalview Authors. (University of Dundee, Dundee, UK.)
715 2014 Genomes C


Gagarine Yaikhom (Medical Research Council Harwell, Oxfordshire, Harwell OX11 0RD, United Kingdom)
717 2014 Other C

Jun Seita, Debashis Sahoo, David L. Dill, Irving L. Weissman (Stanford University, Stanford, CA, USA)
718 2014 Transcripts C

Bálint Antal, Anatole Chessel, Rafael E. Carazo Salas (University of Cambridge, UK)
719 2014 Populations C

Artur Wolny (Nencki Institute of Experimental Biology, Pasteur Street 3, 02-093 Warsaw, Poland)
720 2014 Other C

Corinna Vehlow, Daniel Weiskopf (Universität Stuttgart, Visualisierungsinstitut (VISUS), Allmandring 19, 70569 Stuttgart)
721 2014 Other C

Michel Westenberg (Eindhoven University of Technology)
722 2014 Other C

Marco Biasini, Stefan Bienert, Andrew Waterhouse, Konstantin Arnold, Tobias Schmidt, Lorenza Bordoli,
Gabriel Studer, Tiziano Gallo Cassarino, Torsten Schwede (Swiss Institute of Bioinformatics, Biozentrum, Universität Basel, Switzerland)
723 2014 Proteins C

Suzanne Paquette, Kalle Leinonen, William Longabaugh (Institute for Systems Biology, Seattle WA USA)
724 2014 Genomes C

Björn Sommer, Tobias Hoppe, Christian Bender (Bio-/Medical Informatics Department, Bielelefeld University, Bielefeld, Germany)
725 2014 Cells C

Marti Bernardo Faura, Thomas Cokealer, Ioannis Melas, Julio Saez-Rodriguez (EMBL-EBI. Wellcome Trust Genome Campus. CB10 1SD, Hinxton, UK)
726 2014 Populations C

Nikolaus Sonnenschein and Markus Herrgard (Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kogle Allé 6, DK-2970 Hørsholm, Denmark )
727 2014 Genomes C

Mike Karampelias, Ricardo Tejos, Tomasz Nodzynski, Jiri Friml, Steffen Vanneste (VIB - Department of Plant Systems Biology, Ghent University - 9052 Ghent, Belgium;)
728 2014 Proteins C

Radoslaw Suchecki, Ute Baumann (Australian Centre for Plant Functional Genomics, University of Adelaide, PMB 1 Glen Osmond South Australia 5064)
729 2014 Genomes C

Christoph Gille (Institut for Biochemistry, Charite, Berlin)
730 2014 Transcripts C

Fathiah Zakham (Hodeidah University, Faculty of Medicine and Health Sciences)
731 2014 Genomes D

Alexandra Pina Kingman, Anne-Kristin Stavrum, Ivan Viola, Helwig Hauser (Thormøhlensgate 55, 5020 Bergen Norway)
732 2014 Other D

Dr Markus Schmidt, Camillo Meinhart, Dr Olga Radchuk (Kundmanngasse 39/12, 1030 Vienna, Austria)
733 2014 Genomes D

Stefan Simm, Mario Selymesi, Mario Keller, Enrico Schleiff (Department of Biosciences, Molecular Cell Biology of Plants Goethe University, Frankfurt am Main, Germany)
734 2014 Genomes D

The sbv Improver project team (Phillip Morris Products SA, Research and Development, Neuchâtel, Switzerland)
735 2014 Genomes D

Katrine Kirkeby Skeby (a), Emad Tajkhorshid (b) and Birgit Schiøtt (a) (a.Department of Chemistry, iNANO, and inSPIN centers, Aarhus University, b. Department of Biochemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois.)
736 2014 Proteins D

Julian Heinrich, Michael Krone, Sean O'Donoghue, Daniel Weiskopf (CSIRO, Sydney, Australia; Visualization Research Center, Stuttgart, Germany; Garvan Institute of Medical Research, Sydney, Australia)
737 2014 Proteins D

Cedric C. Laczny, Patrick May, Nikos Vlassis, Paul Wilmes (Luxembourg Centre for Systems Biomedicine, Esch-sur-Alzette, Luxembourg)
738 2014 Genomes D

Daniel Gerighausen, Dirk Zeckzer, Lydia Steiner, Sonja J. Prohaska (Institut für Informatik, Universität Leipzig, Härtelstr. 16-18, D-04107 Leipzig)
739 2014 Other D

Leyla Garcia, Xavier Watkins, Maria J Martin (EMBL-EBI, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, United Kingdom)
740 2014 Proteins D

Dharani Dhar Burra, Therese Bengtsson, Estelle Proux Wera, Erik Andreasson, Erik Alexandersson (Department of plant protection biology, Swedish University of Agricultural Sciences, Alnarp, Sweden, SE-23053)
741 2014 Transcripts D

E. Wait, M. Winter, C. Bjornsson, S. Goderie, S. Temple, and A. Cohen (Drexel University, Philadelphia, PA; Neural Stem Cell Institute, Rensselaer, NY)
742 2014 Cells D

Nico Scherf, Michael Weber, Michaela Mickoleit, Benjamin Schmid, Jan Huisken (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany)
743 2014 Cells D

Wolfram Liebermeister1, Elad Noor2, Dan Davidi2, Avi Flamholz2, Ron Milo2, Katharina Riedel3, Henry Mehlan3, Julia Schüler4, Jörg Bernhardt3 (1Charité Berlin, 2Weizmann Institute of Science, Greifswald University 3Institute of Microbiology, 4Institute for Mathematics )
744 2014 Organisms D

Julia Schüler1, Jörg Bernhardt2, Andreas Spillner1, Katharina Riedel2 (1 Institute of Mathematics, Greifswald, Germany; 2 Institute of Microbiology, Greifswald, Germany)
745 2014 Organisms D

Nadezhda T. Doncheva, John H. Morris, Karsten Klein, Eric F. Pettersen, Michael Wybrow, Conrad C. Huang, Francisco S. Domingues, Thomas E. Ferrin, Mario Albrecht (Max Planck Institute for Informatics, Saarbruecken; University of California, San Francisco; University of Sydney; Monash University, Melbourne; EURAC research, Bolzano; University Medicine Greifswald)
746 2014 Proteins D

Derek Wright, Tim Angus and Tom C. Freeman (The Roslin Institute, The University of Edinburgh, Easter Bush, Midlothian EH25 9RG. Scotland)
747 2014 Cells D


Hector Zenil, Venkateshan Kannan, Jesper Tegnér (Karolinska Institutet)
749 2014 Genomes B

Katrine Kirkeby Skeby (a), Emad Tajkhorshid (b) and Birgit Schiøtt (a) (a.Department of Chemistry, iNANO, and inSPIN centers, Aarhus University, b. Department of Biochemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois.)
750 2014 Art & Biology Y

Nick R. Love (RIKEN: Center for Developmental Biology, Kobe, Japan)
751 2014 Art & Biology Y

Marcin Klapczynski (Private Work)
752 2014 Art & Biology Y


William J.R. Longabaugh (Institute for Systems Biology, Seattle WA USA)
754 2014 Art & Biology Y


Cedric C. Laczny, Patrick May, Nikos Vlassis, Paul Wilmes (Luxembourg Centre for Systems Biomedicine, Esch-sur-Alzette, Luxembourg)
756 2014 Art & Biology Y

Beat Wolf(1), Pierre Kuonen(1), Thomas Dandekar(2) ((1)University of Applied Sciences Western Switzerland, Fribourg, Switzerland; (2)University of Würzburg, Würzburg, Germany)
757 2014 Art & Biology Y

Ryo Sakai, Peter Konings, Yves Moreau, Koen Vanmechelen, Jan Aerts (KU Leuven, Leuven, Belgium; iMinds Medical IT, Leuven, Belgium)
758 2014 Art & Biology Y

Alexandra Pina Kingman, Anne-Kristin Stavrum, Ivan Viola, Helwig Hauser (Thormøhlensgate 55, 5020 Bergen Norway)
759 2014 Art & Biology Y

E. Wait, M. Winter, C. Bjornsson, S. Goderie, S. Temple, and A. Cohen (Drexel University, Philadelphia, PA; Neural Stem Cell Institute, Rensselaer, NY)
760 2014 Art & Biology Y

Heng-Chang Chen (Centre for Genomic Regulation (CRG), Edif. PRBB, Dr. Aiguader, 88, 08003, Barcelona, Spain )
761 2014 Art & Biology Y

Paula González Avalos, Lucas Schütz, Silvia Urbansky, Stefan Günther, Heike Leitte, Steffen Lemke (Im Neuenheimer Feld 230)
762 2014 Art & Biology Y

Christopher Hammang (CSIRO, 11 Julius Avenue, North Ryde, Sydney, NSW, 2113)
763 2014 Art & Biology Y

Julia Schüler (Institute of Mathematics and Computer Science; Greifswald; Germany)
764 2014 Art & Biology Y

Wencke Walter, Carlos Torroja, Fatima Sanchez-Cabo, Mercedes Ricote (Centro Nacional Investigaciones Cardiovasculares (CNIC) Carlos III, Madrid, Spain)
765 2014 Art & Biology Y

Oscar Reina (IRB Barcelona. C/Baldiri Reixac 10. 08028, Barcelona. Spain)
766 2014 Art & Biology Y

1Jörg Bernhardt, 2Frank Schmidt (Greifswald University 1Institute of Microbiology, 2Interfaculty Institute for Genetics and Functional Genomics)
767 2014 Art & Biology Y

Kenneth Sabir (Garvan Institute, Sydney Australia)
768 2014 Art & Biology Y
")
Christian Stolte, Christopher Hammang (CSIRO Computational Informatics, Sydney, Australia)
769 2014 Art & Biology Y

Seán O'Donoghue; Jim Procter; Bang Wong (CSIRO & Garvan Inst.; U. Dundee; Broad Inst.)
770 2013 Art & Biology Y

Ivan Cao-Berg, Aabid Shariff, Jieyue Li, Devin Sullivan, Tao Peng, Armaghan Naik, Gustavo Rohde, Robert F. Murphy (Lane Center for Computational Biology, Carnegie Mellon University, Pittsburgh, Pennsylvania, USA)
771 2013 Cells A

William J.R. Longabaugh (Institute for Systems Biology, Seattle, WA USA)
772 2013 Other A

Ryo Sakai, Matthieu Moisse, Joke Reumers, Jan Aerts (KU Leuven, Leuven, Belgium)
773 2013 Genomes A

Thomas Abeel, Thomas Van Parys, Ashlee Earl, Bruce Birren, Yves Van de Peer (Broad Institute, Cambridge, USA;Ghent University, Ghent, Belgium)
774 2013 Genomes A

EKATERINA GALKINA, SHWETA PURUSHE, AND GEORGES G. GRINSTEIN (1 University Ave, Lowell, MA)
775 2013 Populations A

Michaela Spitzer, Juri Rappsilber, Mike Tyers (Wellcome Trust Centre for Cell Biology, Edinburgh, UK; IRIC, Montreal, Canada)
776 2013 Other A

A Heilbut, X Zhu, P Tamayo, ME Hatten, E Kolaczyk, JP Mesirov (7 Cambridge Center, Cambridge, MA)
777 2013 Transcripts A

Eric Pettersen, Conrad Huang, Tom Goddard, Graham Johnson, Greg Couch, David Mischel, Scooter Morris, Elaine Meng, and Thomas Ferrin (University of California, San Francisco; 600 16th Street; San Francisco, CA 94158-2517; USA)
778 2013 Proteins A

L. Unson, C. Neves, C. Allan, J. Swedlow (Glencoe Software, Inc. Seattle, WA)
779 2013 Other B

Xin Zhou, Ting Wang (Washington University in St. Louis, St. Louis, USA)
780 2013 Genomes B

William Moore, Chris Allan, Jason Swedlow et.al (Wellcome Trust Centre for Gene Regulation & Expression, University of Dundee, Dundee, Scotland, UK; LOCI, University of Wisconsin-Madison, Madison, WI, USA; Glencoe Software, Inc.)
781 2013 Genomes B

Mikhail Chernykh, Yannis Kalaidzidis, Giovanni Marsico, Claudio Collinet, Nikolay Samusik, Thierry Galvez, Marino Zerial (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany)
782 2013 Genomes B

William Ray, Christopher Bartlett, Raghu Machiraju (Nationwide Children's Hospital, Columbus Ohio, USA; The Ohio State University, Columbus Ohio, USA)
783 2013 Proteins B

Kate Patterson, Sean O'Donoghue, Drew Berry, Christopher Hammang, Maja Divjak, Lidija Bosnjak, Christian Stolte (Garvan Institute, 384 Victoria Street Darlinghurst NSW 2010)
784 2013 Other B

Dan Gurnon, Tom Goddard, Jacob Stanley, Julian Voss-Andreae, Dave Berque, Paul Fesenmeir (DePauw University, Greencastle, IN, USA; University of California San Francisco, CA, USA)
785 2013 Other B

Brian Craft, Teresa Swatloski, Mary Goldman, Kyle Ellrott, Melissa Cline, Christopher Wilks, Singer Ma, Christopher Szeto, Josh Stuart, David Haussler, Jingchun Zhu (UC Santa Cruz, Santa Cruz, CA, USA)
786 2013 Genomes C

Cagatay Turkay, Julius Parulek, Helwig Hauser (University of Bergen, Bergen, Norway)
787 2013 Genomes C

Bang Wong, Aravind Subramanian, Josh Gould, Corey Flynn, Todd Golub, and the CMap team (Broad Institute, 7 Cambridge Center, Cambridge, MA 02142, USA)
788 2013 Transcripts C

Graham Johnson with Autodesk (University of California, San Francisco (qb3@UCSF))
789 2013 Cells C

Qiaonan Duan, Edward Y. Chen, Christopher M. Tan, Aravind Subramanian, Mario Niepel, Peter K. Sorger, Avi Ma’ayan (Icahn School of Medicine at Mount Sinai, New York, NY, USA)
790 2013 Transcripts C

Alberto I. Roca (ProfileGrid.org, Irvine, CA, USA)
791 2013 Populations C

Nacho Caballero (Boston University)
792 2013 Transcripts D

Katherine Celler, Roman I. Koning, Gilles P. van Wezel (Molecular Biotechnology, Institute of Biology, Leiden University, Leiden, Netherlands; Electron Microscopy, Molecular Cell Biology, Leiden University Medical Centre, Leiden, Netherlands)
793 2013 Organisms D

James Rosindell (Imperial College London)
794 2013 Other D

Michael Krone, Martin Falk, Thomas Ertl (Visualization Research Center, University of Stuttgart, Germany)
795 2013 Proteins D

Kenneth Sabir, Sean O’Donoghue, Christian Stolte, Nelson Perdigão, Vivian Ho, Venkata P. Satagopam, Maria Kalemanov, David Ma, Burkhard Rost, Andrea Schafferhans (CSIRO (Australia), Garvan Inst. (Australia), TUM (Germany), Tech. U. Lisbon (Portugal), U. Luxembourg (Luxembourg) U. Sydney (Australia))
796 2013 Proteins D

Xin-Yi Chua, James M. Hogan, Sean Chalmers, Amber Weightman, Xixin Zhou (School of EECS, QUT, Brisbane Australia)
797 2013 Transcripts D

Christopher Hammang, Sean O'Donoghue, Christian Stolte, Drew Berry, Trevor Lockett, Julie Clarke, Leah Cosgrove, David Topping (11 Julius Ave, North Ryde, Sydney, NSW, Australia)
798 2013 Cells D

Henry Mehlan, Michael Hecker, Katharina Riedel, Jörg Bernhardt (Greifswald University, Jahnstr. 15, 17487 Greifswald; DECODON GmbH, Rathenau-Str. 49a, 17489 Greifswald; Germany)
799 2013 Populations E

Günter Jäger, Florian Battke, Karsten Borgwardt, Kay Nieselt (Center for Bioinformatics Tübingen, University of Tübingen, Germany; Max-Planck-Institute for Intelligent Systems, Tübingen, Germany )
800 2013 Transcripts E

Oliver Schwengers, Tim. W. Nattkemper, Anke Becker, Alexander Goesmann (Center for Biotechnology, Bielefeld, Germany)
801 2013 Other E

Gideon Dresdner, Jianjiong Gao, Bülent Arman Aksoy, Ugur Dogrusoz, Benjamin Gross, S. Onur Sumer, Yichao Sun, Anders Jacobsen, Rileen Sinha, Erik Larsson, Ethan Cerami, Chris Sander, Nikolaus Schultz (1275 York Ave, New York, NY)
802 2013 Genomes E

Maciek Smuga-Otto, Michele Arduengo, Isobel Maciver (Promega Corporation, Madison WI, USA)
803 2013 Cells E

Christian Stolte, Lars J Jensen, Heiko Horn, Sune Pretscher-Frankild, Kenneth Sabir, Seán I O’Donoghue (Garvan Institute of Medical Research, Sydney, CSIRO CMIS, Sydney, NNF Center for Protein Research, University of Copenhagen, Denmark. University of Sydney, Australia)
804 2013 Other E

Christoph Wolf, Daniel Erkan, Arash Nasir Tafreshi, Werner Baumgartner (Department for Cellular Neurobionics, Institute for Biology 2, RWTH-Aachen University, Aachen, Germany)
805 2013 Cells A

Petri Klemelä, Aleksi Kallio, Taavi Hupponen, Massimiliano Gentile, Jarno Tuimala, Ari-Matti Saren, Rimvydas Naktinis, Eija Korpelainen (CSC - IT Center for Science Ltd., P.O. Box 405, FI-02101 Espoo, Finland)
806 2013 Genomes B

Derek Wright, Tim Angus and Tom C. Freeman (The Roslin Institute, The University of Edinburgh, Easter Bush, Midlothian EH25 9RG. Scotland, UK)
807 2013 Cells C

Imtiaz Khan, Adam Fraser, Mark-Anthony Bray, Paul Smith, Anne Carpenter, Rachel Errington (School of Medicine, Cardiff University, UK; Broad Institute of MIT and Harvard, Cambridge, USA)
808 2013 Other D

Felice Frankel (MIT, USA)
809 2013 Art & Biology Y

Christoph Wolf, Daniel Erkan, Arash Nasir Tafreshi, Werner Baumgartner (Department for Cellular Neurobionics, Institute for Biology 2, RWTH-Aachen University, Aachen, Germany)
810 2013 Art & Biology Y

William J.R. Longabaugh (Institute for Systems Biology, Seattle, WA USA)
811 2013 Art & Biology Y

Mikhail Chernykh, Yannis Kalaidzidis, Giovanni Marsico, Claudio Collinet, Nikolay Samusik, Thierry Galvez, Marino Zerial (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany)
812 2013 Art & Biology Y

Henry Mehlan, Michael Hecker, Katharina Riedel, Jörg Bernhardt (Greifswald University, Jahnstr. 15, 17487 Greifswald; DECODON GmbH, Rathenau-Str. 49a, 17489 Greifswald; Germany)
813 2013 Art & Biology Y

Michael Krone, Martin Falk, Thomas Ertl (Visualization Research Center, University of Stuttgart, Germany)
814 2013 Art & Biology Y

Maciek Smuga-Otto, Brian Rosenberg, Keith V. Wood, Monika G. Wood, Thomas Machleidt, and Matt B. Robers (Promega Corporation, Madison, WI, USA)
815 2013 Art & Biology Y

Dan Gurnon (DePauw University, Greencastle, IN, USA)
816 2013 Art & Biology Y

Christopher Hammang, Sean O'Donoghue, Christian Stolte, Drew Berry, Trevor Lockett, Julie Clarke, Leah Cosgrove, David Topping (11 Julius Ave, North Ryde, Sydney, NSW, Australia)
817 2013 Art & Biology Y

William Ray (Nationwide Children's Hospital, Columbus Ohio, USA)
818 2013 Art & Biology Y


Kate Patterson (Garvan Institute, 384 Victoria Street Darlinghurst NSW 2010)
820 2013 Art & Biology Y


Fabian de Kok-Mercado (HHMI, Dept. of Science Education, Chevy Chase, MD)
822 2013 Art & Biology Y

Seán O'Donoghue, Jim Procter (EMBL Heidelberg, Germany)
823 2012 Art & Biology Y

Chandan Sona and V Badireenath Konkimalla (National Institute of Science Education and Research, Institute of Physics Campus, Sachivalaya Marg, PO: Sainik School, Bhubaneswar - 751 005 India)
824 2012 Genomes A

Matheus Regis Belisário, Adriano Marcelo Morgon, Luiza Helena Gremski, Waldemiro Gremski, Andréia Senff Ribeiro, Olga Meiri Chaim, Silvio Sanches Veiga. (Federal University of Paraná, Curitiba, Brazil.)
825 2012 Proteins A

Markus-Hermann Koch; Julian Uszkoreit, Stefan Loroch, Katja Kuhlmann, Martin Eisenacher, Hanna Diehl, Raphael Voges, Marco Oldiges, Helmut E. Meyer, Christian Stephan (MPC, Universitaetsstrasse 150 (ZKF I), 44801 Bochum, Germany)
826 2012 Other A

Roman Koning (P.O.Box 9600, 2300 RC, Leiden, The Netherlands)
827 2012 Cells A

Corinna Vehlow, Jan Hasenauer, Andrei Kramer, Julian Heinrich, Nicole Radde, Frank Allgöwer and Daniel Weiskopf (Visualisierungsinstitut, Allmandring 19, 70569 Stuttgart)
828 2012 Other A

Malcolm Hinsley, Gemma Barton, Ed Griffths ((Wellcome Trust Sanger Institute, Genome Campus, Hinxton, Cambridge, CB10 1SA, UK)
829 2012 Genomes A

Naamah Bloch, David Harel (Weizmann Institute of Science, Rehovot, Israel)
830 2012 Cells A

Sebastian Bremm, Martin Heß, Kay Hamacher, Tatiana von Landesberger (Graphisch-Interaktive Systeme, TU Darmstadt, Fraunhoferstr. 5, 64283 Darmstadt, Germany; Bioinformatics and theo. Biology, TU Darmstadt, Schnittspahnstr. 10, 64287 Darmstadt, Germany;)
831 2012 Organisms A

Jeremy Henty, Gemma Barson (Wellcome Trust Sanger Institute, Genome Campus, Hinxton, Cambridge)
832 2012 Genomes A

Alain Meil and Patrick Durand (Korilog - 4, rue Gustave Eiffel - 56230 Questembert - France - info@korilog.com)
833 2012 Genomes A

Julia Dmitrieva and Laurence Lins (Centre de Biophysique Moléculaire Numérique Gembloux Agro-Bio Tech, Université de Liège, Gembloux, Belgium)
834 2012 Cells A

Michael P Schroeder, Christan Pérez-Llamas, Abel Gonzalez-Perez & Nuria Lopez-Bigas (Research Unit on Biomedical Informatics, Universitat Pompeu Fabra, Dr. Aiguader 88, 08003 Barcelona, Spain)
835 2012 Genomes A

C Anderson, SD Harris, EN moriyama (University of Nebraska, Lincoln, Nebraska)
836 2012 Genomes A

Daria B. Kokh, Stefan Henrich, Paul Czodrowski, Friedrich Rippmann, Rebecca C. Wade (Heidelberg Institute for Theoretical Studies (HITS gGmbH), Schloss-Wolfsbrunnenweg 35, D-69118 Heidelberg, Germany)
837 2012 Proteins A

M.P. van Iersel, E.J. Gonçalves, C. Chaouiya, J. Saez-Rodriguez (EMBL-EBI, Wellcome Trust Genome Campus, Cambridge, UK)
838 2012 Cells A

Josh Moore, Jason R. Swedlow, and the OME Consortium (Wellcome Trust Centre for Gene Regulation & Expression, University of Dundee, Dundee, Scotland, UK)
839 2012 Other A

S. Simon, D. Oelke, R. Landstorfer, K. Neuhaus and D. A. Keim (Universitätsstraße 10, Box 78, 78457 Konstanz)
840 2012 Transcripts A

Sarah-Jane Schramm1, Anna E Campain1, Simone S Li2, David C Y Fung2, Vivek Jayaswal1, Chi Nam Ignatius Pang2, Richard A Scolyer1&3&4, Yee Hwa Yang1, Marc R Wilkins2 and Graham J Mann1&3 (1The University of Sydney, Sydney, Australia; 2University of New South Wales, Sydney, Australia; 3Melanoma Institute Australia, Sydney Australia; 4Royal Prince Alfred Hospital, Sydney, Australia )
841 2012 Transcripts A

Christian Stolte & Sean O’Donoghue (CSIRO Mathematics, Informatics & Statistics, Sydney, Australia)
842 2012 Proteins A

Kubra Karagoz, Kazım Yalçın Arga (Department of Bioengineering, Marmara University, Goztepe 34722, Istanbul, Turkey )
843 2012 Cells B

M. Kamran Azim, Yong Zhang, M. Iqbal Choudhary, Zhixiang Yan, Chuanchun Yang, Asifullah Khan, Huma Asif and Yasmeen Rashid (BGI, Shenzhen, China; 2PCMD, University of Karachi, Karachi, Pakistan)
844 2012 Genomes B

Georgios A. Pavlopoulos, Alejandro Sifrim, Ryo Sakai, Jan Aerts (Katholieke Universiteit Leuven, Depatment of Electrical Engineering (ESAT), Leuven, Belgium)
845 2012 Genomes B

M. Chavent, A. Tek, Z. Lu, M. Piuzzi, B. Laurent, M. Baaden (1: Laboratoire de Biochimie Théorique, CNRS, UPR9080, Univ Paris Diderot, Sorbonne Paris Cité, Paris, France. 2: SBCB Unit, Department of Biochemistry, University of Oxford, Oxford, United Kingdom.)
846 2012 Proteins B

Marco Biasini, Konstantin Arnold, Stefan Bienert, Andrew Waterhouse, Torsten Schwede (Biozentrum, University of Basel, Klingelbergstrasse 50 / 70, 4056 Basel, Switzerland)
847 2012 Proteins B

Mark M. Shovman, James Bown (University of Abertay, Dundee, UK)
848 2012 Cells B


S. P. Gore, G. van Ginkel, S. Velankar, G. J. Kleywegt (EBI, Cambridge, United Kingdom)
850 2012 Proteins B

Johannes Wollnik (Genomatix Software GmbH, Bayerstr. 85a, 80335 Munich, Germany)
851 2012 Transcripts B

Christian Panse, Jonas Grossmann, Bertran Gerrits, Ralph Schlapbach (Functional Genomics Center Zurich)
852 2012 Genomes B

Vladyslav Luzin, Vladimir Ovcharenko, Anton Yeryomin (Lugansk state medical university, Lugansk, Ukraine)
853 2012 Organisms B

Marc Piuzzi, Alex Tek, Matthieu Chavent, Nicolas Férey, Matthieu Dréher, Ahmed Turki, Sébastien Limet, Sophie Robert, Bruno Raffin, Marc Baaden (Institut de Biochimie des protéines, Paris, France; CEA/DIF, Bruyères le Châtel, France; INRIA-MOAIS,Grenoble, France; LIFO, Orléans, France)
854 2012 Proteins B

Colin Combe; Salman Tahir; Lutz Fischer; Zhuo Angel Chen; Juan Zou; Adam Belsom; Lauri Peil; Jimi-Carlo Bukowski-Wills; Juri Rappsilber (Wellcome Trust Centre for Cell Biology, King's Buildings, University of Edinburgh, Mayfield Road, Edinburgh, Scotland EH9 3JR )
855 2012 Proteins B

Christoph Kuehne, Frank Depoix, Jürgen Markl (Institute of Zoology, Johannes Gutenberg University, Mainz, Germany)
856 2012 Proteins B

Marc Fiume, Andrew Brook, Eric Smith, and Michael Brudno (Department of Computer Science, University of Toronto, Toronto, Canada)
857 2012 Genomes B

Maik Röder, David González and Roderic Guigó (Centre for Genomic Regulation (CRG) and UPF, Dr. Aiguader 88, 08003 Barcelona, Spain)
858 2012 Transcripts B

Renate Matzke-Karasz, Radka Symonova, Libor Morkovsky (Faculty of Science, Charles University in Prague, Czech Republic; Department of Earth and Environmental Sciences, Ludwig-Maximilians-University Munich, Germany)
859 2012 Organisms B

Evan Molinelli, Martin Miller, Arman Aksoy (Computational Biology Center, Memorial Sloan Kettering Cancer Center, New York, NY USA)
860 2012 Cells B

Katy Borner & Michael J. Stamper (Curators) (Indiana University)
861 2012 Other B

Adriano M. Morgon, Matheus R. Belisário, Gabriel O. Meissner, Larissa Vuitika, Dilza Trevisan-Silva, Andrea S. Ribeiro, Luiza H. Gremski, Olga M. Chaim, Waldemiro Gremski, Silvio S. Veiga (Federal University of Parana, Curitiba, Brazil)
862 2012 Proteins C

Maria Kokkinopoulou and Jürgen Markl (Johannes Gutenberg Universität Mainz, Johannes-von-Müller-Weg 6, 55099, Mainz, Germany)
863 2012 Cells C

Peter Droste, Wolfgang Wiechert, Katharina Nöh (IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany)
864 2012 Other C

Oksana Riba-Grognuz, Laurent Keller, Laurent Falquet, Ioannis Xenarios and Yannick Wurm (Department of Ecology and Evolution, University of Lausanne, Lausanne, Switzerland; Swiss Institute for Bioinformatics, University of Lausanne, Lausanne, Switzerland)
865 2012 Genomes C

Michael Kohl, Maike Ahrens, Hagen Meckel, Julian Uszkoreit, Michael Turewicz, Martin Eisenacher, Dominik A. Megger, Barbara Sitek, Helmut E. Meyer, Christian Stephan (Medizinisches Proteom-Center, Ruhr-Universitaet Bochum, Bochum, Germany)
866 2012 Other C

Samuel Hertig & Viola Vogel (Laboratory for Biologically Oriented Materials, HCI Building, Wolfgang-Pauli-Strasse 10, 8093 Zürich)
867 2012 Proteins C

Ingvar Lagerstedt, Ardan Patwardhan, Gerard Kleywegt (EBI, Cambridge, United Kingdom)
868 2012 Proteins C

M. F. Symmons, S. P. Gore, M. J. Conway, G. van Ginkel, S. Velankar, G. J. Kleywegt (EBI, Cambridge, United Kingdom)
869 2012 Proteins C

Trevor Paterson, Martin Graham, Jessie Kennedy, Andy Law (Edinburgh Napier University & The Roslin Institute, University of Edinburgh)
870 2012 Genomes C

Cagatay Turkay, Julius Parulek, Helwig Hauser (University of Bergen, Bergen, Norway)
871 2012 Proteins C

Malte Mader, Ronald Simon, Sascha Steinbiss, Stefan Kurtz (Center for Bioinformatics, University of Hamburg, Hamburg, Germany; Institute of Pathology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany)
872 2012 Genomes C

Günter Jäger, Florian Battke, Corinna Vehlow, Julian Heinrich, Kay Nieselt (University of Tübingen, Germany; University of Stuttgart, Germany;)
873 2012 Other C

João PGLM Rodrigues, Mikaël Trellet, Christophe Schmitz, Adrien SJ Melquiond, Alexandre MJJ Bonvin (Padualaan 8, Utrecht, The Netherlands)
874 2012 Proteins C

Heiko Stark, Rosemarie Fröber, Nadja Schilling (Institut für Spezielle Zoologie und Evolutionsbiologie, Friedrich-Schiller-Universität Jena, Deutschland; Institut für Anatomie I, Friedrich-Schiller-Universität Jena, Deutschland)
875 2012 Organisms C

Zhian Lv, Alex Tek, Matthieu Chavent, Marc Baaden (Laboratoire de Biochimie Théorique - IBPC, Paris, France)
876 2012 Proteins C

Sven Haag, Daniel Nowak, W.-K. Hofmann, Markus Gumbel (Department of Hematology and Oncology, University Medical Centre Mannheim, Germany; Institute for Medical Informatics, University of Applied Sciences Mannheim, Germany)
877 2012 Genomes C

Björn Sommer, Yan Zhou (Bio-/Medical Informatics Department, Bielefeld University, Universitätsstraße 25, 33615 Bielefeld, Germany)
878 2012 Cells C

Thanos Theo and Tom Freeman (The Roslin Institute The University of Edinburgh Easter Bush Midlothian EH25 9RG Scotland, UK)
879 2012 Transcripts C

Khushbu Kumari and Kishore CS Panigrahi (NATIONAL INSTITUTE OF SCIENCE EDUCATION AND RESEARCH, SCHOOL OF BIOLOGICAL SCIENCE, BHUBANESWAR ,ODISHA, 751005, INDIA)
880 2012 Other C

Peter Sisk, Christian Stolte, Thomas Abeel, Matthew Peterson, Jeremy Zucker, Antonio Gomes, Anna Lyubetskaya, Tige Rustad, Kyle Minch, Irina Glotova, David Sherman, James Galagan (Broad Institute of MIT and Harvard, Cambridge, MA; Boston University, Boston, MA; Seattle Biomed, Seattle, WA)
881 2012 Genomes D

Jens Fangerau, Burkhard Höckendorf, Jochen Wittbrodt, Bernhard X. Kausler, Fred A. Hamprecht, Heike Leitte (Interdisciplinary Center for Scientific Computing, Heidelberg, Germany; Centre for Organismal Studies, Heidelberg, Germany; Heidelberg Collaboratory for Image Processing, Heidelberg, Germany)
882 2012 Organisms D

Maria Secrier, Reinhard Schneider (European Molecular Biology Laboratory, Heidelberg, Germany)
883 2012 Other D

Megan Riel-Mehan, Kevan Shokat (600 16th St San Francisco CA 94158)
884 2012 Cells D

Claudia Pommerenke, Tatiana Nedelko, Klaus Schughart (Helmholtz-Center for Infection Research, Infection Genetics, Braunschweig, Germany)
885 2012 Genomes D

Leonid Zaslavsky, Vyacheslav Chetvernin, Boris Fedorov, Boris Kiryutin, Alexandre Souvorov, Igor Tolstoy, and Tatiana Tatusova (National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland 20894, USA)
886 2012 Genomes D

Michaela Blazikova, Ivan Novotny, Petr Herman, Jan Malinsky (Faculty of Mathematics and Physics, Charles University, Prague, Czech Republic;Institute of Experimental Medicine, ASCR, Prague; Institute of Molecular Genetics, ASCR, Prague)
887 2012 Genomes D

Petri Klemelä, Aleksi Kallio, Taavi Hupponen, Massimiliano Gentile , Rimvydas Naktinis, Eija Korpelainen (CSC - IT Center for Science Ltd., P.O. Box 405, FI-02101 Espoo, Finland)
888 2012 Genomes D

Sabine C. Mueller, Stefan Nickels, D. Stoeckel, A.K. Dehof, H.P. Lenhof, Andreas Hildebrandt (Intel Visual Computing Institute, Center for Bioinformatics, Saarbrücken, Saarland, Johannes-Gutenberg Universität, Mainz)
889 2012 Proteins D

Ryo Sakai, Matthieu Moisse, Joke Reumers, Alejandro Sifrim, Raf Winand, Georgios A. Pavlopoulos, Andrew Vande Moere, Jan Aerts (Department of Electrical Engineering (ESAT-SCD), KU Leuven, Belgium; IBBT Future Health Department, Belgium)
890 2012 Genomes D

S. Venkataraman, Bill Hill, Zsolt Husz, Chris J. Armit, Dominic Rannie, Richard A. Baldock (MRC Human Genetics Unit, MRC IGMM, University of Edinburgh, Western General Hospital, Crewe Road South, Edinburgh EH4 2XU, UK)
891 2012 Organisms D

Tobias Czauderna and Falk Schreiber (IPK Gatersleben & MLU Halle-Wittenberg, Germany)
892 2012 Cells D

Oliver Staehlin, Stephanie Perreau-Lenz, Wolfgang Sommer (Central Institute of Mental Health, Mannheim, Germany)
893 2012 Transcripts D

Roy Ruddle, Darren Treanor & A Johannes Pretorius (University of Leeds, Leeds, UK)
894 2012 Other D


Martin Falk, Thomas Ertl (Visualization Research Center, University of Stuttgart)
896 2012 Cells D

Alexander Lex, Marc Streit, Hans-Joerg Schulz, Christian Partl, Dieter Schmalstieg, Peter J. Park, Nils Gehlenborg (Harvard Medical School, Boston, USA; Broad Institute, Cambridge, USA; Graz University of Technology, Graz, Austria; Johannes Kepler University, Linz, Austria; University of Rostock, Rostock, Germany)
897 2012 Other D

Ludovic Autin, Graham Johnson, Johan Hake, Arthur Olson, Michel Sanner (10550 N. Torrey Pines Road)
898 2012 Proteins D


Seán O'Donoghue, Jim Procter, Larry Hunter, Bang Wong (EMBL, Germany; U. Dundee, UK; U. Colorado Denver, USA, Broad, USA)
900 2011 Art & Biology X

Drew Berry (Walter & Eliza Hall, Australia )
901 2011 Art & Biology X

Larry Kryala, Florian Block, Tanja Gesell, Hanspeter Pfister, and Chia Shen (Harvard University)
902 2011 Genomes A

David Kidd (Imperial College London, Ascot, United Kingdom)
903 2011 Other A

Victor Chistyakov, Mick Correll, John Quackenbush (Dana-Farber Cancer Institute, Boston, MA 02215, USA )
904 2011 Genomes A

Kate Patterson and Susan Clark (Garvan Institute of Medical Research, 384 Victoria Street Darlinghurst, NSW, Australia, 2010)
905 2011 Genomes A

Annika Kreuchwig, Gunnar Kleinau, Franziska Kreuchwig, Catherine L. Worth, Gerd Krause (Leibniz-Institut für Molekulare Pharmakologie, Robert-Rössle-Str. 10, 13125 Berlin, Germany)
906 2011 Proteins A

Georg Tremmel, Atsushi Niida, Satoru Miyano (Human Genome, Center Institute of Medical Science University of Tokyo 4-6-1 Shirokanedai, Minatoku, Tokyo 108-8639, Japan)
907 2011 Genomes A

Saraswathi Abhiman (NCBI, 8600 Rockville Pike, Bethesda MD, 20894 USA)
908 2011 Proteins A

Martin Falk, Thomas Ertl (VISUS - Visualization Research Center, University of Stuttgart, Germany)
909 2011 Cells A


Aurora Cain, Adam Price, Shatavia S. Morrison,
Robert Kosara, Cynthia Gibas (University of North Carolina at Charlotte)
911 2011 Genomes A

Oliver SP Davis (King's College London, London, UK)
912 2011 Populations A

Jennifer A. Frazier, Joyce Ma, Isaac Liao, Kwan Liu Ma (Exploratorium, 3601 Lyon Street, San Francisco, CA 94123)
913 2011 Populations A

Shantanu SIngh, Raghu Machiraju, Thierry Pecot, Gustavo Leone (395 Dreese Laboratories, 2015 Neil Avenue, Columbus, OH 43016)
914 2011 Cells A

Carsten Görg, Hannah Tipney, Karin Verspoor, William A Baumgartner Jr., K. Bretonnel Cohen, John Stasko, Lawrence Hunter (Center for Computational Pharmacology, University of Colorado Denver School of Medicine, USA; School of Interactive Computing, Georgia Institute of Technology, USA)
915 2011 Other B

Michel A. Westenberg & Jos B.T.M. Roerdink (Department of Mathematics and Computer Science, Eindhoven University of Technology, Netherlands; Johann Bernoulli Institute for Mathematics and Computer Science, University of Groningen, Netherlands.)
916 2011 Transcripts B

M. Chavent, A. Tek, Z. Lu, M. Baaden (Laboratoire de Biochimie Théorique)
917 2011 Proteins B

Megan Strait, Connor Gramazio, Jisoo Park, Sara Su, Lenore Cowen (Tufts University, Medford, MA, USA)
918 2011 Proteins B

Kay Nieselt, Albert Pritzkau, Andreas Lehrmann (Center for Bioinformatics Tübingen, Tübingen, Germany; )
919 2011 Transcripts B

Elham Gharazi, Albert Y. Zomaya (School of Information Technologies, J12 University of Sydney NSW 2006 Australia)
920 2011 Other B

Xin Zhou, Mingchao Xie, Cydney Nielsen, Martin Hirst, Peggy Farnham, Robert Kuhn, Jingchun Zhu, Jim Kent, David Haussler, Joseph Costello, Ting Wang (1. Washington University in St. Louis, 2. BC Cancer Agency, 3. University of Southern California, 4. University of California at Santa Cruz, 5. University of California San Francisco)
921 2011 Genomes B

Chris North, Alex Endert, Ankit Singh, Robert Kincaid (Virginia Tech, Blacksburg, USA; Agilent Technologies, Santa Clara, USA)
922 2011 Transcripts B

Kozo Nishida (Nara institute of science and technology, Ikoma, Japan)
923 2011 Cells B

Raluca Andrei, Stefano Cianchetta, Tiziana Loni, Maria Francesca Zini and Monica Zoppè (scientifici Visualization Unit, IFC - CNR. Pisa italy)
924 2011 Proteins B

Chase A. Miller, Michelle M. Meyer (140 Commonwealth Avenue, Chestnut Hill, MA 02467)
925 2011 Genomes B

Surendra kumar, Ralf Neumann and Kamran Shalchian-Tabrizi (Department of Biology, P.O. Box 1066 Blindern, Oslo, Norway)
926 2011 Genomes B

Marc Fiume, Eric Smith, Andrew Brook, Vanessa Williams, Michael Brudno (University of Toronto , 6 King's College Rd, Toronto, ON M5S 3G4, Canada)
927 2011 Genomes B

Georgios A. Pavlopoulos, Maria Secrier, Reinhard Schneider, Jan Aerts (Katholieke Universiteit Leuven, Faculty of Engineering - ESAT/SCD, Kasteelpark Arenberg, 3001 Leuven-Heverlee, Belgium)
928 2011 Other C
 microscope for diagnostic pathology")
Roy Ruddle, Darren Treanor, Rebecca Randell, Rhys Thomas & Phil Quirke (University of Leeds, Leeds, England)
929 2011 Other C

Britta Weber, Marit Möller, Steffen Prohaska, David Günther, Hans-Christian Hege (Zuse Institute Berlin, Berlin, Germany)
930 2011 Genomes C

Damien Lariviere, Eric Fourmentin, Ines Winkler, Peter Friedhoff, Terence Strick (Fourmentin-Guilbert Scientific Foundation, Noisy-Le-Grand, France; Institute for Biochemistry, FB 08, Justus Liebig University, Giessen, Germany; Institut Jacques Monod, CNRS UMR 7592, Paris, France)
931 2011 Proteins C

Daniel Gusenleitner, John Quackenbush, Aedin Culhane (Dana Farber Cancer Institute, Boston, USA)
932 2011 Transcripts C

Lawrence Buckingham, Xin-Yi Chua, James M. Hogan (MQUTeR, Queensland University of Technology, GPO Box 2434, Brisbane QLD 4001, AUSTRALIA)
933 2011 Genomes C

Keiichiro Ono (University of California, San Diego, USA)
934 2011 Cells C

Markus Schröder, Daniel Gusenleitner, Alexander Goesmann, Aedín Culhane, John Quackenbush, Benjamin Haibe-Kains (Biostatistics and Computational Biology, Dana-Farber Cancer Institute, Boston, USA)
935 2011 Transcripts C

Martin Gütlein, Andreas Karwath, Stefan Kramer (Albert-Ludwigs-Universität Freiburg, Germany; Technische Universität München, Germany)
936 2011 Genomes C

M Krzywinski, I Birol, S Jones, M Marra (Canada’s Michael Smith Genome Sciences Centre, Vancouver, Canada)
937 2011 Genomes C

Thomas Abeel, James Galagan, Yves Van de Peer (Broad Institute of MIT and Harvard, Cambridge, MA, USA; Ghent University, Gent, Belgium)
938 2011 Genomes C

Karen Cranston, Adam Kubach, Kristopher Urie (National Evolutionary Synthesis Center, Durham, USA; Texas Advanced Computing Center, Austin, USA; Biodiversity Synthesis Center, Chicago, USA)
939 2011 Populations C

S Venkataraman, Bill Hill, Zsolt Husz, Ruben Schilling1, Alexander Schliep2, Lorna Richardson, Jeff Christiansen, Duncan Davidson & Richard Baldock (MRC Human Gentics Unit, Institute of Genetics & Molecular Medicine, Western General Hospital, Crewe Road South, Edinburgh EH4 2XU, UK)
940 2011 Organisms C

Daniel Pick, Nao Hisakawa, Forest Rohwer (Computational Science Research Center, San Diego State University, San Diego, CA 92182)
941 2011 Genomes D

A. Johannes Pretorius, Roy A. Ruddle (University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK)
942 2011 Genomes D

Danielle Albers, Colin Dewey, and Michael Gleicher (University of Wisonsin - Madison, 1210 W Dayton St., Madison, WI;)
943 2011 Genomes D

Julian Heinrich, Corinna Vehlow, Kay Nieselt, Florian Battke, Daniel Weiskopf (Visualization Research Center (VISUS), Stuttgart, Germany; Integrative Transcriptomics, Tübingen, Germany)
944 2011 Genomes D

Alex Lomsadze, Mark Borodovsky (Georgia Institute of Technology, Atlanta, GA, USA)
945 2011 Genomes D

Imtiaz Khan, Adam Fraser, Mark Bray, Victoria Griesdoorn, Paul Smith, Anne Carpenter, Rachel Errington (Cardiff University, Cardiff, UK & Broad Institute of MIT and Harvard, Cambridge, USA)
946 2011 Cells D

Christian A. Grove, Paul W. Sternberg (California Institute of Technology, 1200 E California Blvd, Pasadena, CA 91125)
947 2011 Cells D

Jan Huisken (Max Planck Institute of Molecular Cell Biology)
948 2011 Organisms D

Alberto I. Roca, Aaron C. Abajian, David J. Vigerust (University of California, Irvine, USA)
949 2011 Populations D

Jörg Bernhardt; Henry Mehlan; Julia Schüler; Michael Hecker (E.M.A.-University Greifswald, Institute for Microbiology; Jahnstrasse15;17487 Greifswald; Germany)
950 2011 Transcripts D

William Ray (Nationwide Children's Hospital, Columbus Ohio, USA; The Ohio State University, Columbus Ohio, USA)
951 2011 Other D

Megan Pirrung, Ryan Kennedy, Rob Knight (University of Colorado at Boulder)
952 2011 Genomes D

John "Scooter" Morris, Thomas Ferrin (University of California San Fancisco, San Francisco, CA, USA)
953 2011 Proteins D

954 2011 Art & Biology X


Marc Baaden, Chantal Prévost (Laboratoire de Biochimie Théorique, Paris, France)
956 2011 Art & Biology X

Marc Baaden (Laboratoire de Biochimie Théorique, Paris, France)
957 2011 Art & Biology X

Matthieu Chavent, Marc Baaden (Laboratoire de Biochimie Théorique, Paris, France)
958 2011 Art & Biology X

Damien Lariviere, Eric Fourmentin, Ines Winkler, Peter Friedhoff, Terence Strick (Fourmentin-Guilbert Scientific Foundation, Noisy-Le-Grand, France; Institute for Biochemistry, FB 08, Justus Liebig University, Giessen, Germany; Institut Jacques Monod, CNRS UMR 7592, Paris, France)
959 2011 Art & Biology X

Kate Patterson (Garvan Institute of Medical Research, 384 Victoria Street Darlinghurst, NSW, Australia, 2010)
960 2011 Art & Biology X

Lawrence Buckingham, Xin-Yi Chua, James M. Hogan (MQUTeR, Queensland University of Technology, GPO Box 2434, Brisbane QLD 4001, AUSTRALIA)
961 2011 Art & Biology X
")
Keiichiro Ono (University of California, San Diego, USA)
962 2011 Art & Biology X

Christian A. Grove (California Institute of Technology, 1200 E California Blvd, Pasadena, CA 91125)
963 2011 Art & Biology X

Matthieu Chavent, Marc Baaden, Syma Khalid (Laboratoire de Biochimie Théorique, Paris, France and Systems and Synthetic Biology Modelling Group, Southampton, United Kingdom)
964 2011 Art & Biology X

Tiziana Loni, Stefano Cianchetta and Monica Zoppè (scientifici Visualization Unit, IFC - CNR. Pisa italy)
965 2011 Art & Biology X

Saraswathi Abhiman (NCBI, 8600 Rockville Pike, Bethesda MD, 20894 USA)
966 2011 Art & Biology X

M Krzywinski, I Birol, S Jones, M Marra (Canada’s Michael Smith Genome Sciences Centre, Vancouver, Canada)
967 2011 Art & Biology X


Tiziana Loni, Stefano Cianchetta and Monica Zoppè (scientifici Visualization Unit, IFC - CNR. Pisa italy)
969 2011 Art & Biology X

Lawrence Buckingham, Xin-Yi Chua, James M. Hogan (MQUTeR, Queensland University of Technology, GPO Box 2434, Brisbane QLD 4001, AUSTRALIA)
970 2011 Art & Biology X

William Ray (Nationwide Children's Hospital, Columbus Ohio, USA; The Ohio State University, Columbus Ohio, USA)
971 2011 Art & Biology X

Shareef Dabdoub and William Ray (Nationwide Children's Hospital, Columbus Ohio, USA; The Ohio State University, Columbus Ohio, USA)
972 2011 Art & Biology X

Kirk Moldoff (Kirk Moldoff Illustration, Princeton, USA)
973 2011 Art & Biology X

Kirk Moldoff (Kirk Moldoff Illustration, Princeton, USA)
974 2011 Art & Biology X



Kirk Moldoff (Kirk Moldoff Illustration, Princeton, USA)
977 2011 Art & Biology X

Larry Kryala, Florian Block, Tanja Gesell, Hanspeter Pfister, and Chia Shen (Harvard University)
978 2011 Art & Biology X

Sean O'Donoghue, Jim Procter, and Nils Gehlenberg (EMBL Heidelberg, Germany)
979 2010 Art & Biology X

Bang Wong (Broad Institute, USA)
980 2010 Art & Biology X

Keiichiro Ono (University of California, San Diego, La Jolla, United States of America)
981 2010 Cellular Systems F

Simon Brent (Wellcome Trust Sanger Institute, Cambridge, United Kingdom)
982 2010 Genomes F

Donovan Parks (Dalhousie University, Halifax, Canada)
983 2010 Populations F

Rainer Pielot (Leibniz-Institute for Neurobiology, Magdeburg, Germany)
984 2010 Cellular Systems F

John Pinney (Imperial College London, London, United Kingdom)
985 2010 Genomes F

Leighton Pritchard (Scottish Crop Research Institute, Dundee, United Kingdom)
986 2010 Genomes F

Albert Pritzkau (ICCAS, Leipzig, Germany)
987 2010 Transcripts F

Steffen Prohaska (Zuse Institute Berlin (ZIB), Berlin, Germany)
988 2010 Cellular Systems F

Ermir Qeli (Institute of Molecular Biology, University of Zurich, Zurich, Switzerland)
989 2010 Transcripts F

Graham Ritchie (Wellcome Trust Sanger Institute, Cambridge, United Kingdom)
990 2010 Genomes F

Hendrik Rohn (Institute of Plant Genetics and Crop Plant Research Gatersleben, Gatersleben, Germany)
991 2010 Cellular Systems F

Stephan Saalfeld (Max Planck Institute of Molucular Cell Biology and Genetics, Dresden, Germany)
992 2010 Cellular Systems F

Athina Samara (BRFAA, Athens, Greece)
993 2010 Cellular Systems F

Annapaola Santarsiero (EMBL-EBI, Cambridge, United Kingdom)
994 2010 Transcripts F

Andrea Schafferhans, Seán O'Donoghue (Technische Universitaet Muenchen, Garching, Germany; EMBL Heidelberg, Germany)
995 2010 Proteins F

Johannes Schmid (Med. Univ. Vienna, Vienna, Austria)
996 2010 Proteins F

Enrico Bertini (University of Konstanz, Konstanz, Germany)
997 2010 Cellular Systems F

Björn Sommer (Bielefeld University, Bielefeld, Germany)
998 2010 Cellular Systems F

Stephan Symons (University of Tuebingen, Tuebingen, Germany)
999 2010 Transcripts F

Stephen Taylor (Weatherall Institute of Molecular Medicine, Oxford, United Kingdom)
1000 2010 Genomes F

Alex Tek (CNRS - IBPC, Paris, France)
1001 2010 Proteins F

Lieven Thorrez (Katholieke Universiteit Leuven, Leuven, Belgium)
1002 2010 Transcripts F

Hannah Tipney (University of Colorado at Denver Health Sciences Center, Aurora, United States of America)
1003 2010 Cellular Systems F

Marjan Trutschl (Louisiana State University, Shreveport, United States of America)
1004 2010 Cellular Systems F

Mariette van de Corput (Erasmus Medical Center Rotterdam, Rotterdam, Netherlands)
1005 2010 Proteins F

Huub van de Wetering (Technische Universiteit Eindhoven, Eindhoven, Netherlands)
1006 2010 Genomes F

Klaas Vandepoele (VIB - Ghent University, Ghent, Belgium)
1007 2010 Genomes F

Michel Westenberg (Eindhoven University of Technology, Eindhoven, Netherlands)
1008 2010 Transcripts F

Monica Zoppè (Scientific Visualization Unit - Inst. of Clinical Physiology, Pisa, Italy)
1009 2010 Proteins F

Christoph Gille (Humboldt University, Berlin, Germany)
1010 2010 Populations F

Salvador Santiago (EMBL, Heidelberg, Germany)
1011 2010 Proteins F

Anna Hupalowska (International Institute of Molecular and Cell Biology, Warsaw, Poland)
1012 2010 Cellular Systems T

Matthew Huska (MDC Berlin, Berlin, Germany)
1013 2010 Populations T

Gregory Jordan (EMBL-EBI, Cambridge, United Kingdom)
1014 2010 Genomes T

Maria Jordao (INSA, Lisbon, Portugal)
1015 2010 Transcripts T
: a graphical tool to study structured RNA genes")
Fabrice Jossinet (University of Strasbourg, Strasbourg, France)
1016 2010 Populations T

Laszlo Kajan (Technische Universitat Munchen, Garching, Germany)
1017 2010 Populations T

Aleksi Kallio (CSC, The IT Center for Science, Espoo, Finland)
1018 2010 Transcripts T

Rikke Kjaergaard (MRC Mitochondrial Biology Unit, Cambridge, United Kingdom)
1019 2010 Proteins T

Petri Klemelä (CSC, The IT Center for Science, Espoo, Finland)
1020 2010 Genomes T

Kim Kristiansen (Glostrup Research Institute, Glostrup, Denmark)
1021 2010 Cellular Systems T

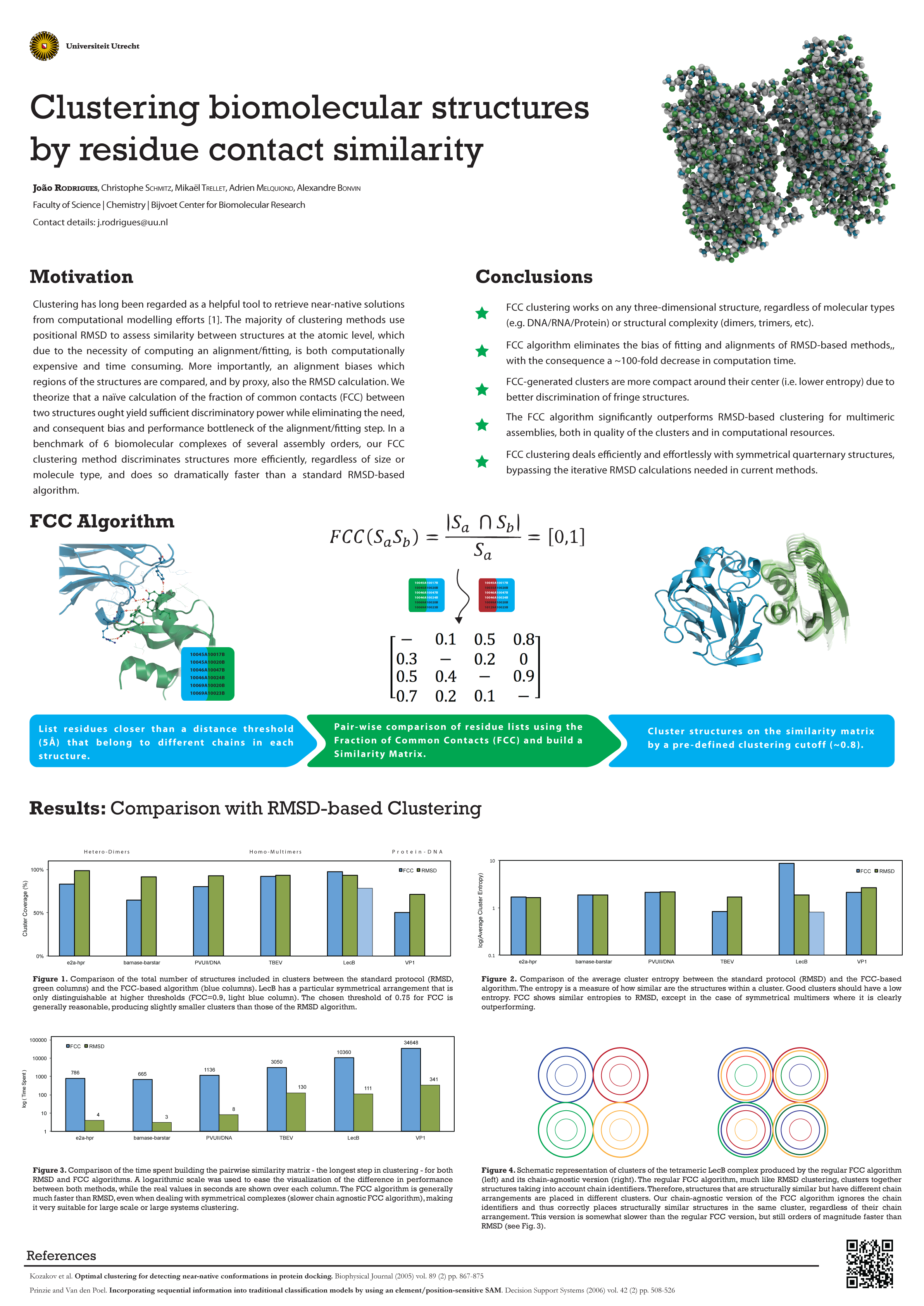{kind=link}

Srinivas Kudavelly (Philips Research Asia - Bangalore, Bangalore, India)
1023 2010 Genomes T

Damien Larivière (Fourmentin-Guilbert Scientific Foundation, Noisy-Le-Grand, France)
1024 2010 Proteins T

David Leader (University of Glasgow, Glasgow, United Kingdom)
1025 2010 Proteins T

Wen Hwa Lee (SGC & University of Oxford, Oxford, United Kingdom)
1026 2010 Proteins T

Frederic Fol Leymarie (Goldsmiths, University of London, London, United Kingdom)
1027 2010 Proteins T

Juliane Liepe (Imperial College London, London, United Kingdom)
1028 2010 Cellular Systems T

Victor Renault (CEPH Fondation Jean Dausset, Paris, France)
1029 2010 Genomes T

Melanie Lou (McMaster University, Hamilton, Canada)
1030 2010 Populations T

Dan MacLean (The Sainsbury Laboratory, Norwich, United Kingdom)
1031 2010 Genomes T

Lukas Marsalek (Saarland University, Saarbruecken, Germany)
1032 2010 Proteins T

Miriah Meyer (Harvard University, Cambridge, United States of America)
1033 2010 Transcripts T

Sébastien Moretti (University of Lausanne, Lausanne, Switzerland)
1034 2010 Populations T

Hiroyuki Nakamura (Keio University, Kanagawa, Japan)
1035 2010 Transcripts T
 2D Plot")
Goran Neshich (EMBRAPA Informatica Agropecuaria, Campinas, Brazil)
1036 2010 Proteins T
: Insights into the relationship between hydrophobic effect and oligomerization")
Izabella Neshich (UNICAMP, Campinas, Brazil)
1037 2010 Proteins T

Heiko Neuweger (Bielefeld University, Bielefeld, Germany)
1038 2010 Transcripts T

Harm Nijveen (Wageningen University, Wageningen, Netherlands)
1039 2010 Transcripts T

Daniela Oelke (University of Konstanz, Konstanz, Germany)
1040 2010 Populations T

Per Oksvold (Royal Institute of Technology, Stockholm, Sweden)
1041 2010 Transcripts T

Kliment Olechnovic (Institute of Biotechnology, Vilnius, Lithuania)
1042 2010 Proteins T

Andrew Flaus (NUI Galway, Galway, Ireland)
1043 2010 Proteins T

Thomas Abeel (Ghent University, Gent, Belgium)
1044 2010 Genomes W

Carolina Agop-Nersesian (University Heidelberg, Medical School, Heidelberg, Germany)
1045 2010 Cellular Systems W

Nadezhda Doncheva (Max Planck Institute for Informatics, Saarbruecken, Germany)
1046 2010 Proteins W

Maria Secrier (EMBL, Heidelberg, Germany)
1047 2010 Transcripts W

Christoph Best (EMBL-EBI, Cambridge, United Kingdom)
1048 2010 Proteins W

Christophe Blanchet (CNRS IBCP, Lyon, France)
1049 2010 Populations W

Matthieu Chavent (CNRS, Paris, France)
1050 2010 Proteins W
: A visual interface for the exploratory analysis of microarray time-course data")
Paul Craig (Edinburgh Napier University, Edinburgh, United Kingdom)
1051 2010 Transcripts W

Rute da Fonseca (CIIMAR - Center for Marine & Environmental Research, Porto, Portugal)
1052 2010 Proteins W

Oliver Davis (King's College London, London, United Kingdom)
1053 2010 Transcripts W

Bernard de Bono (EMBL-EBI, Cambridge, United Kingdom)
1054 2010 Transcripts W

Victor de la Torre (Spanish National Cancer Research Centre, Madrid, Spain)
1055 2010 Transcripts W

Franck Debarbieux (IBDML, Marseille, France)
1056 2010 Cellular Systems W

Michael Deyholos (University of Alberta, Edmonton, Canada)
1057 2010 Genomes W

Pablo Echeverria (UNIGE, Geneva, Switzerland)
1058 2010 Transcripts W

Venkata Satagopam (EMBL, Heidelberg, Germany)
1059 2010 Transcripts W

Dorothea Emig (Max Planck Institute for Informatics, Saarbruecken, Germany)
1060 2010 Transcripts W

Geoffrey Fucile (University of Toronto, Toronto, Canada)
1061 2010 Transcripts W

Nils Gehlenborg (EMBL-EBI, Cambridge, United Kingdom)
1062 2010 Transcripts W

Tanja Gesell (Max F. Perutz Laboratories (MFPL), Vienna, Austria)
1063 2010 Proteins W

Christoph Gille (Humboldt University, Berlin, Germany)
1064 2010 Populations W

Carsten Goerg (Georgia Institute of Technology, Atlanta, United States of America)
1065 2010 Transcripts W

Christian Grove (California Institute of Technology, Arcadia, United States of America)
1066 2010 Cellular Systems W

Sven Haag (EMBL, Heidelberg, Germany)
1067 2010 Transcripts W

Stephan Hegge (Heidelberg University, Heidelberg, Germany)
1068 2010 Cellular Systems W

Julian Heinrich (University of Stuttgart, Stuttgart, Germany)
1069 2010 Transcripts W

Des Higgins (Conway Institute, Dublin, Ireland)
1070 2010 Populations W

James Hogan (Queensland University of Technology, Brisbane, Australia)
1071 2010 Populations W

Andreas Hoppe (Charité Berlin, Berlin, Germany)
1072 2010 Transcripts W

Jan Huisken (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany)
1073 2010 Cellular Systems W

Lawrence Hunter (University of Colorado School of Medicine, Aurora, United States of America)
1074 2010 Transcripts W

Cydney Nielsen (BC Cancer Agency Genome Sciences Centre, Vancouver, Canada)
1075 2010 Genomes W

Stefan Suhrer (University of Salzburg, Salzburg, Austria)
1076 2010 Proteins X